一种基于几何网络的蛋白质结构预测方法与流程
1.本发明涉及生物信息学、计算机应用领域,进一步的,涉及蛋白质三级结构预测领域,尤其涉及,一种基于几何网络的蛋白质结构预测方法。
背景技术:
2.蛋白质结构预测是生命科学领域的一大难题。生命所必需的每一次基础生物学进展几乎都是由蛋白质带来的。蛋白质参与创建细胞和组织并保持这它们的形状,构成维持生命所需化学反应的催化酶,充当分子工场,转运工具和马达,充当细胞通讯的信号和接收器等等。
3.蛋白质有很多氨基酸长链组成,通过折叠成精确的3d结构来完成无数的任务,这些结构控制着他们与其他分子互动的方式。蛋白质由20种不同的氨基酸组成。这些氨基酸就像字母表中的字母,组合成单词、句子、段落,产生了无数种可能的文本。然而,与字母不同,氨基酸是位于3d空间的物理存在。通常,蛋白质的各个部分在物理上非常接近,但在序列上却相隔很远,因为其氨基酸链形成了环、螺旋、折叠等不同形状。
4.蛋白质的形状决定了其功能以及它在疾病中的功能紊乱程度。阐明蛋白质的结构是所有分子生物学的核心,更是治疗患者、拯救生命、改变生活的医学发展的核心。
5.近年来,根据氨基酸序列预测蛋白质折叠结构方面的计算方法已经取得了很大进展。如果能够充分实现,这些方法可能会改变生物医学研究的方方面面。然而,现在的方法在可测定的蛋白质的大小和范围上是有限的。虽然成功率高,但利用物理工具来鉴别蛋白质结构的过程既昂贵又耗时,即使是使用现代技术(如低温电子显微镜)同样如此。因此,绝大多数蛋白质结构以及致病突变对这些结构的影响目前仍是未知的。蛋白质折叠方式的计算方法有可能大大降低确定蛋白质结构的成本和时间。但经过近四十年的不懈努力,这个难题仍未解决。
6.因此,本领域迫切需要一种成本低、费时少的蛋白质结构预测方法。
技术实现要素:
7.本发明的目的在于,提供一种基于几何网络的蛋白质结构预测方法,基于端到端的可微分深度学习方法,具有成本低且费时少的优点。
8.本发明通过氨基酸序列的表达式和pssm矩阵信息,经过循环几何网络直接得到蛋白质的扭转角信息,进而得到蛋白质的三级结构。本技术人在此基础上完成了本发明。
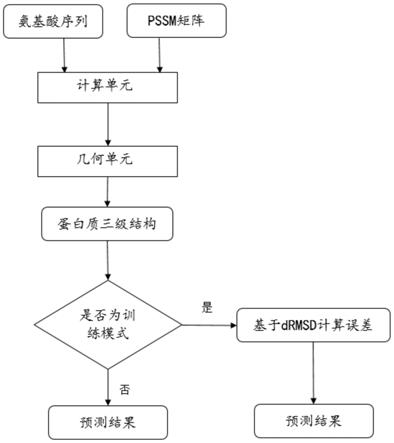
9.一种基于几何网络的蛋白质结构预测方法,所述方法包括步骤:
10.s1,将一个氨基酸序列和pssm矩阵(位置特异性打分矩阵)输入到计算单元,得到计算单元的隐状态;
11.s2,将计算单元的隐状态输入几何单元,几何单元计算给定残基的扭转角和由其上游的几何单元产生的部分完成的骨干,得到一个残基延伸的新骨干,该骨干被输入至相邻的下游单元,得到蛋白质的扭转角信息,最后输出蛋白质的完整三级结构;
12.s3,基于drmsd(距离的均方根误差)度量来计算预测结构和实验结构之间的误差。
13.在一些实施方式中,所述计算单元由循环几何网络构成,对于每个氨基酸残基位置(residue position),计算单元将氨基酸信息和pssm矩阵信息与来自相邻单元的信息进行整合,得到计算单元的隐状态。
14.进一步的,对每个氨基酸残基的计算整合了从残基上游和下游的信息,覆盖了整个蛋白质。
15.进一步的,计算单元为gru cell、或lstm cell中的一种。
16.进一步的,gru cell拟合更快,适用于参数量少、数据量较小的情况下,lstm cell拟合相对较慢,适用于参数量多、数据量较大的情况下。
17.进一步的,本发明中,所述计算单元优选为gru cell。
18.进一步的,所述gru cell的核心部件为双向门控循环单元(bi-gru)。
19.进一步的,所述gru cell的表达式为公式m1:
20.z
t
=σ(w
z
·
[h
t-1,
x
t
])
[0021]
r
t
=σ(w
r
·
[h
t-1
,x
t
])
[0022][0023][0024]
其中,wz、wr和w为计算单元参数,x
t
为集成了氨基酸序列和pssm矩阵信息的向量,h
t
为计算单元的隐状态。
[0025]
进一步的,为了更好地利用输入信息,在本发明的计算单元为两个gru cell,分别为前向gru cell和反向gru cell,两个gru cell架构完全一致,参数独立学习。
[0026]
进一步的,对于每个gru cell,将关于氨基酸信息和pssm矩阵信息与来自相邻单元的信息进行整合,再通过循环双向拓扑结构,对每个残基的计算整合了从残基上游和下游的信息,覆盖了整个蛋白质。
[0027]
在一些实施方式中,在步骤s2中,计算单元的隐状态h
t
输入几何单元的计算公式为m2:
[0028][0029][0030]
其中,wφ为几何单元模型参数,bφ是一个偏差向量,φ是一个待学习的字母表,φt为最终输出的三个扭转角。
[0031]
在一些实施方式中,在步骤s3中,所述drmsd首先计算预测结构中所有原子和实验结构中所有原子的成对距离,然后计算这些距离集合之间的均方根。
[0032]
进一步的,在训练模式下,需要步骤s3,基于drmsd计算误差,然后预测结果;不在训练模式下,不需要再经过步骤s3,直接预测结果。
附图说明
[0033]
图1为本发明的整体流程方框图。
[0034]
如下将结合具体实施案例对附图进行具体说明。
具体实施方式
[0035]
描述以下实施例以辅助对本发明的理解,实施例不是也不应当以任何方式解释为限制本发明的保护范围。
[0036]
在以下描述中,本领域的技术人员将认识到,在本论述的全文中,组件可描述为单独的功能单元(可包括子单元),但是本领域的技术人员将认识到,各种组件或其部分可划分成单独组件,或者可整合在一起(包括整合在单个的系统或组件内)。
[0037]
此外,附图内的组件或系统之间的连接并不旨在限于直接连接。相反,在这些组件之间的数据可由中间组件修改、重格式化、或以其它方式改变。另外,可使用另外或更少的连接。还应注意,术语“联接”、“连接”、或“输入”应理解为包括直接连接、通过一个或多个中间设备来进行的间接连接、和无线连接。
[0038]
实施例1:
[0039]
为使本发明的目的、技术方案和优点更加清楚明白,以下结合具体实施例,并参照附图,为本发明作进一步的详细说明。
[0040]
一种基于几何网络的蛋白质结构预测方法,所述方法包括步骤:
[0041]
s1,将一个氨基酸序列和pssm矩阵(位置特异性打分矩阵)输入到计算单元,得到计算单元的隐状态;
[0042]
所述计算单元由循环几何网络构成,对于每个氨基酸残基位置(residueposition),计算单元将氨基酸信息和pssm矩阵信息与来自相邻单元的信息进行整合,得到计算单元的隐状态;对每个氨基酸残基的计算整合了从残基上游和下游的信息,覆盖了整个蛋白质。
[0043]
计算单元为两个gru cell,分别为前向gru cell和反向gru cell,两个gru cell架构完全一致,参数独立学习。所述gru cell的表达式为公式m1:
[0044]
z
t
=σ(w
z
·
[h
t-1,
x
t
])
[0045]
r
t
=σ(w
r
·
[h
t-1
,x
t
])
[0046][0047][0048]
其中,wz、wr和w为计算单元参数,x
t
为集成了氨基酸序列和pssm矩阵信息的向量,h
t
为计算单元的隐状态;
[0049]
对于每个gru cell,将关于氨基酸信息和pssm矩阵信息与来自相邻单元的信息进行整合,再通过循环双向拓扑结构,对每个残基的计算整合了从残基上游和下游的信息,覆盖了整个蛋白质。
[0050]
s2,将计算单元的隐状态输入几何单元,几何单元计算给定残基的扭转角和由其上游的几何单元产生的部分完成的骨干,得到一个残基延伸的新骨干,该骨干被输入至相
邻的下游单元,得到蛋白质的扭转角信息,最后输出蛋白质的完整三级结构。
[0051]
进一步的,计算单元的隐状态h
t
输入几何单元的计算公式为m2:
[0052][0053][0054]
其中,wφ为几何单元模型参数,bφ是一个偏差向量,φ是一个待学习的字母表,φt为最终输出的三个扭转角。
[0055]
s3,基于drmsd(距离的均方根误差)度量来计算预测结构和实验结构之间的误差。所述drmsd首先计算预测结构中所有原子和实验结构中所有原子的成对距离,然后计算这些距离集合之间的均方根。然后预测结果。
[0056]
尽管本发明已公开了多个方面和实施方式,但是其它方面和实施方式对本领域技术人员而言将是显而易见的,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。本发明公开的多个方面和实施方式仅用于举例说明,其并非旨在限制本发明,本发明的实际保护范围以权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1