基于深度学习的白细胞抗原与多肽结合亲和力预测方法与流程
本发明涉及免疫治疗和人工智能技术领域,特别涉及一种基于深度学习的白细胞抗原与多肽结合亲和力预测方法。
背景技术:
目前,人类白细胞抗原与多肽的结合对于表位多肽的递呈到细胞表面并激活随后的t细胞免疫反应具有非常重要的作用。通过构建机器学习模型来预测人类白细胞抗原和多肽的结合亲和力已经成功被应用于免疫治疗的靶标选择。一般来说人类白细胞抗原与多肽的结合预测可以分为抗原亚型特异和泛抗原亚型两种方法。抗原亚型特异方法需要为每一种人类白细胞抗原亚型构建预测模型。另一方面,泛人类白细胞抗原亚型方法可以通过整合白细胞抗原核心区域进行编码,从而对所有的人类白细胞抗原亚型及多肽进行亲和力预测。在过去的若干年中,人类白细胞抗原与多肽结合的实验数据以及机器学习算法共同提高了结合力的预测精度。然而现有的方法由于实验数据的偏向性(与人类白细胞抗原i型a和b类相比,i型c类的实验数据量相对较少),对于i型c类的预测精度有待进一步提高。同时由于i型人类白细胞抗原结合的多肽的长度为8个氨基酸至15个氨基酸,现有算法对于相对较长的多肽(12到15个氨基酸)的预测精度远次于短多肽。因此开发精度更高的人类白细胞抗原与多肽的结合力预测算法具有重要的临床意义。
技术实现要素:
针对存在的上述问题,本发明综合考虑了人类白细胞抗原蛋白序列与肽段各自的序列对亲和力强度的影响,开发了一种基于深度学习的白细胞抗原与多肽结合亲和力预测方法。
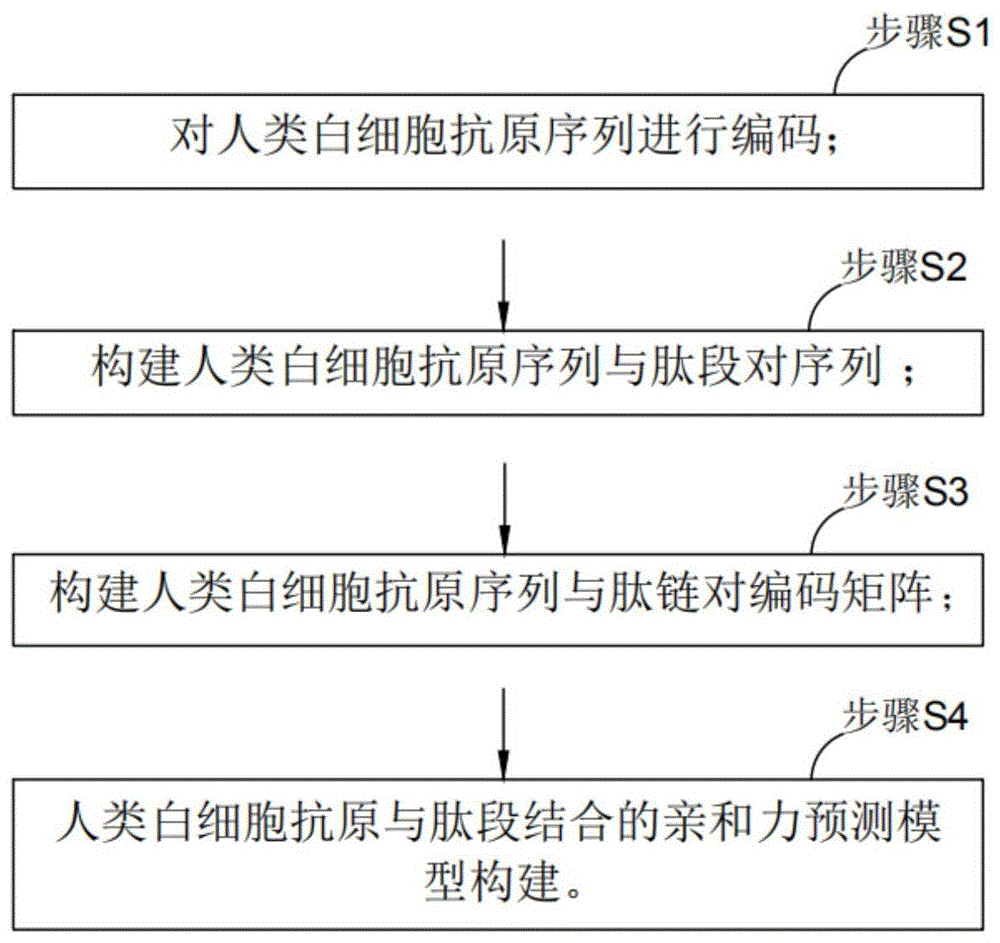
本发明实施例提供的一种基于深度学习的白细胞抗原与多肽结合亲和力预测方法,包括:
步骤s101:对人类白细胞抗原序列进行编码;
步骤s102:构建人类白细胞抗原序列与肽段对序列;
步骤s103:构建人类白细胞抗原序列与肽链对编码矩阵;
步骤s104:人类白细胞抗原与肽段结合的亲和力预测模型构建。
优选的,步骤s104:人类白细胞抗原与肽段结合的亲和力预测模型构建,包括:
步骤s201:捕获编码矩阵序列信息;
步骤s202:从多个角度为氨基酸分配权重;
步骤s203:计算人类白细胞抗原序列-肽段的亲和力。
优选的,步骤s201:捕获编码矩阵序列信息,包括:
将所述编码矩阵序列中的每一个氨基酸视为所述人类白细胞抗原序列中的一个结点;
将所述结点的编码向量依次送入双向长短期记忆网络中;所述双向长短期记忆网络可以分别按照所述编码矩阵序列的正向与逆向顺序对所述编码矩阵序列进行特征学习。
优选的,步骤s202:从多个角度为氨基酸分配权重,包括:
通过多头注意力机制将所述编码矩阵序列的特征映射至多个特征空间,并分别计算在每个特征空间下各氨基酸的注意力权重来量化各氨基酸对人类白细胞抗原序列与肽段关联的重要性。
在多个子空间下,我们能得到每个特征空间下各氨基酸的注意力权重。为了将各个特征空间的权重进行整合,我们还通过一个滤片尺寸为head*1*1的卷积神经网络单独为每个特征空间分配权重,随后分别对于每个氨基酸的多个注意力权重进行加权求和,从而得到序列的重要性向量,公式如下:
w=[w1,w2,...,whead]
其中w为卷积神经网络的滤片矩阵,wh为第h个特征空间所对应的权重,xh为第h个特征空间下各氨基酸的注意力权重向量。
优选的,步骤s203:计算人类白细胞抗原序列-肽段的亲和力,包括:
通过两个全连接层整合特征表示,并利用一个sigmoid函数得到0~1之间的值,作为该对人类白细胞抗原(hla)序列-肽段对的亲和力分值,公式如下:
temp1=tanh(out·w1+b1)
x=sigmoid(temp1·w2+b2)
其中w1与w2分别是两个全连接层的权重矩阵,b1与b2分别为两个全连接层的偏置向量,tanh表示双曲正切变换。
优选的,步骤s101:对人类白细胞抗原序列进行编码,包括:
人类白细胞抗原核心区域的伪序列来表示人类白细胞抗原亚型。
优选的,步骤s102:构建人类白细胞抗原序列与肽段对序列,包括:
将所述伪序列与相应的肽段序列拼接成一个整体,形成长度为42~49的长序列。
优选的,步骤s103:构建人类白细胞抗原序列与肽链对编码矩阵,包括:
采用blosum62矩阵对所述长序列中的每一个氨基酸进行编码,形成维度为lseq*20的所述编码矩阵,其中lseq表示序列的长度;
或,
采用one-hot向量对所述长序列中的每一个氨基酸进行编码,形成所述编码矩阵。
与现有技术相比,本发明的方案具有如下优势:
一、从原理上讲,本发明所利用的深度学习算法能学习到人类白细胞抗原(hla)序列与肽段对更加深层、原始的序列表征,为提供准确可靠的亲和力预测打下坚实的基础。
二、本发明通过基于深度神经网络的双向长短期记忆网络(bidirectionallongshorttermmemorynetwork),通过一个模型即可完成对大多数a类、b类人类白细胞抗原与多种长度肽段的亲和力预测;并且在当前研究数据较少的情况下,对c类人类白细胞抗原与肽段的亲和力预测能达到与前两类一致的稳定性能。实验证实,本算法在a类、b类和c类i型人类白细胞抗原与长度为8~15氨基酸的肽段序列上的预测性能相较其它预测算法而言性能更优,且更加稳定。
三、通过本算法中的多头注意力机制(multipleheadattentionmechanism)从多个角度评估序列中各氨基酸的重要性,最终在预测亲和力强度的时候网络能对序列整体有一个全面的认识,并且有选择的增强或减弱各位点的信息,从而得到更加准确、稳定的亲和力预测结果。同时在此过程中还能展示出序列中各个氨基酸位点对于亲和力强度的贡献大小,从而更加准确的认识与分析它们之间的作用机制。
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在所写的说明书、权利要求书、以及附图中所特别指出的结构来实现和获得。
下面通过附图和实施例,对本发明的技术方案做进一步的详细描述。
附图说明
附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:
图1为本发明实施例中一种基于深度学习的白细胞抗原与多肽结合亲和力预测方法的示意图;
图2为本发明实施例中一种基于深度学习的白细胞抗原与多肽结合亲和力预测方法的算法实现示意图。
具体实施方式
以下结合附图对本发明的优选实施例进行说明,应当理解,此处所描述的优选实施例仅用于说明和解释本发明,并不用于限定本发明。
参见图1和图2所示,为本发明的一个实施例,基于深度学习的白细胞抗原与多肽结合亲和力预测方法包括:
步骤s101:对人类白细胞抗原序列进行编码;
为了便于计算机运算,我们用人类白细胞抗原核心区域的伪序列来表示人类白细胞抗原亚型(http://www.cbs.dtu.dk/services/netmhcpan/)。每一个人类白细胞抗原的伪序列都是长度为34的字符串序列,其中每一个字符代表一个氨基酸。
例如:hla-a*0101的伪序列为“yfamyqenmahtdantlyiiyrdytwvarvyrgy”。
在此步骤中,我们所使用的人类白细胞抗原核心区域的伪序列与肽段序列的组成一致,为后续人类白细胞抗原与肽段序列的拼接与编码提供了方便。
步骤s102:构建人类白细胞抗原序列与肽段对序列;
长度为8~15个氨基酸的肽段被用于后续分析。我们将前一步得到的伪序列与相应的肽段序列拼接成一个整体,形成长度为42~49的长序列,从而用于泛抗原亚型模型的建立。
区别于本领域大多数需要为不同人类白细胞抗原构建多个模型的算法,我们的算法通过一个统一的模型将人类白细胞抗原序列与肽段序列拼接后进行分析,能更全面的考虑到人类白细胞抗原序列与肽段序列的相互关系。因此模型支持的人类白细胞抗原更加宽泛,并且支持日后新发现的人类白细胞抗原,而无需重新训练对应的模型。
步骤s103:构建人类白细胞抗原序列与肽链对编码矩阵;
随后,为了对拼接序列进行运算,我们需要将其进行数字编码。blosum62矩阵是一个在生物信息学中用于序列对比的氨基酸替换打分矩阵,其表示了20种氨基酸之间相互的替换分值。因此我们将blosum62矩阵按行提取出来作为对应氨基酸的特征向量。如氨基酸“y”的blosum62编码为:“-2,-2,-2,-3,-2,-1,-2,-3,2,-1,-1,-2,-1,3,-3,-2,-2,2,7,-1”。随后利用其对前述得到的长序列中的每一个氨基酸进行编码,形成维度为lseq*20的特征编码矩阵,其中lseq表示序列的长度。
替代方案:我们也可以通过one-hot向量对氨基酸进行编码。由于总共涉及到20个氨基酸,因此one-hot编码为长度为20的向量。将氨基酸与向量中的每一位对应,当前氨基酸所在的位为1,其余均为0。假如氨基酸“y”为第19位,那么它的one-hot向量为:“0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,1,0”。
blosum62编码相比较于其它编码方式(如one-hot编码),将携带更多的来自生物背景的知识,能更好的在有限的编码位数中表达出各氨基酸之间的潜在关系。
步骤s104:人类白细胞抗原与肽段结合的亲和力预测模型构建。基于建立的预测模型对人体白细胞抗原与肽段结合的亲和力进行预测;该步骤包括:步骤s201:捕获编码矩阵序列信息;
通过双向长短期记忆网络(bidirectionallongshorttermmemorynetwork)从序列的角度对人类白细胞抗原(hla)序列-肽段编码进行分析。我们将序列中的每一个氨基酸视为序列中的一个结点,随后将各个结点的编码向量依次送入双向长短期记忆网络中。它可以分别按照序列的正向与逆向顺序对序列进行特征学习,这么做的目的是同时捕获序列的上下文特征信息,使得网络能够更好的学习长序列的编码表示。
我们以pytorch框架为例说明网络的学习过程:
首先,给出双向长短期记忆网络(bidirectionallongshorttermmemorynetwork)的定义:
self.lstm=nn.lstm(input_size=parms_net[′len_acid′],
hidden_size=self.hidden_size,
num_layers=self.layer_num,
bidirectional=true)
其中,input_size指定了长序列中氨基酸的长度。size指定了双向长短期记忆网络(bidirectionallongshorttermmemorynetwork)应当使用多大规模的参数分析数据,num_layers指定了所用的网络层数,bidirectional指定使用双向网络(bidirectionallongshorttermmemorynetwork)对数据进行分析。
随后通过outlstm,hiddenlstm=self.lstm(x)获得双向长短期记忆网络(bidirectionallongshorttermmemorynetwork)所学习到的序列特征,其中x为前述编码好的特征矩阵。
以往的人类白细胞抗原与肽段亲和力预测算法需要将不同长度的肽段填充至统一长度再进行预测,这使得计算资源被浪费在大量没有意义的填充字符上。得益于双向长短期记忆网络(bidirectionallongshorttermmemorynetwork)的序列分析特性,我们的算法可以直接支持不同长度的序列分析,在节省计算资源的同时,也使得网络能更加精确的聚焦于序列本身的有效信息上。
步骤s202:从多个角度为氨基酸分配权重;
通过多头注意力机制(multipleheadattentionmechanism)将序列特征映射至多个特征空间,并分别计算在每个特征空间下各氨基酸的注意力权重来量化各氨基酸对人类白细胞抗原(hla)序列与肽段关联的重要性。具体地,通过如下公式实现此过程:
wiatten=hiddenlstm·wiproject
contexti=wiatten·(tanh(outlstm))t
headi=importancei·outlstm
其中,网络先通过若干个投影矩阵wiproject将长短期记忆网络(bidirectionallongshorttermmemorynetwork)中的权重hiddenlstm投影至多个不同的子空间中得到新权重wiatten;outlstm为双向长短期记忆网络(bidirectionallongshorttermmemorynetwork)的输出,经过双曲正切(tanh)函数变换后与wiatten相乘得到上下文向量contexti,表示的是双向序列表示在不同空间下的上下文表达。
为了计算某个角度下原始序列中各氨基酸的重要性,需要先计算所有空间下的上下文向量随后求和,记为total。随后任一空间的上下文向量contexti与total的比值即为各氨基酸在此空间下的重要性,记为importancei。importancei是与序列长度相同向量,其中的每一位表示对应的氨基酸在第i个空间下的重要性,越接近于1表示该氨基酸越重要,而越接近于0表示多头注意力机制(multipleheadattentionmechanism)尝试在第i个空间下屏蔽来自此氨基酸的信息。
最终,原始序列在第i个空间下的加权表示headi就是importancei与双向长短期记忆网络(bidirectionallongshorttermmemorynetwork)的输出outlstm的乘积。根据之前的定义,来自序列重要位置的信息将会被接近1的权重加权,而不重要的位置将会被分配接近0的权重屏蔽。
在多个子空间下,我们能得到若干个不同的加权序列特征表示。为了将各个特征空间的权重进行整合,我们还通过一个滤片为head*1*1卷积神经网络为每个特征空间分配权重,随后分别对每个氨基酸的多个权重进行加权求和,从而得到该氨基酸的重要性,公式如下:
w=[w1,w2,...,whead]
其中w为卷积神经网络的滤片矩阵,wh为第h个特征空间所对应的权重,xh为第h个特征空间下各氨基酸的注意力权重向量。
代码如下:
self.mixhead=nn.conv2d(in_channels=self.head,out_channels=1,kernel_size=1)
importance=self.mixhead(x)
其中in_channels指定卷积的深度与前述的子空间数量一致,out_channels指定卷积的输出深度为1,kernel_size指定滤片的尺寸为1*1,x为多头注意力机制(multipleheadattentionmechanism)的输出。
该步骤不仅聚焦于序列本身,更希望关注那些在序列中起到重要作用的氨基酸。因此我们通过多头注意力机制(multipleheadattentionmechanism)从多个特征空间中对序列各位置的重要性加以评估,并着重关注那些重点位置上的氨基酸信息。从而在不同长度、不同类型的序列上取得一致、稳定的预测性能。
步骤s203:计算人类白细胞抗原序列-肽段的亲和力。
通过两个全连接层整合前述的特征表示,并利用一个sigmoid函数得到0~1之间的值,作为该对人类白细胞抗原(hla)序列-肽段对的亲和力分值,公式如下:
temp1=tanh(out·w1+b1)
x=sigmoid(temp1·w2+b2)
其中w1与w2分别是两个全连接层的权重矩阵,b1与b2分别为两个全连接层的偏置向量。为了增加模型的非线性表达能力,我们还在两个全连接层之间增加一个双曲正切(tanh)变换。sigmoid函数负责将实数转换成0~1之间的小数,表示该对人类白细胞抗原(hla)序列-肽段对的亲和力分值,越接近于1表示亲和力越强。
代码如下:
out_fc1=nh.linear(in_features=2*self.hidden_size,out_features=self.hidden_size)
out_fc2=nn.linear(in_features=self.hldden_slze,out_features=1)
temp1=out_fc1(out)
temp1=torch.tanh(temp1)
temp2=out_fc2(temp1)
x=torch.sigmoid(temp)
如果需要得到具体的亲和力数值,只需要通过对该亲和力分值进行转换即可:
affnity=500001-x
其中x为亲和力分值,affnity为亲和力强度,越接近于0表示亲和力越强。一般而言,500以内的亲和力强度说明该人类白细胞抗原(hla)序列-肽段之间存在较强的亲和力。
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
- 还没有人留言评论。精彩留言会获得点赞!