一种同时检测甲基化水平、基因组变异和插入片段的方法及装置与流程
本发明涉及生物信息学
技术领域:
,具体涉及一种同时检测甲基化水平、基因组变异和插入片段的方法,以及用于实施该方法的装置和设备及相应的计算机可读介质。
背景技术:
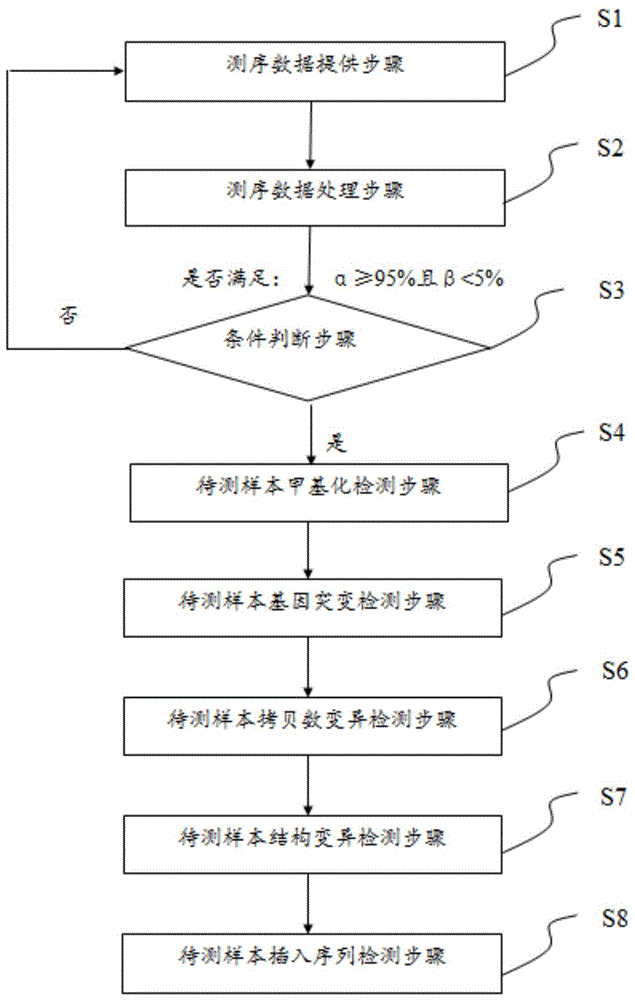
:dna甲基化是dna化学修饰的一种,可以在不改变dna序列的前提下,改变遗传物质。早在1925年,dna甲基化修饰已经被发现。大量研究表明,dna甲基化在基因调控中具有表现遗传作用。dna甲基化中,研究最多的是5-甲基胞嘧啶(5mc),该修饰通常被认为是基因表达的一种稳定的抑制性调控因子。目前基于二代测序技术的dna甲基化检测方法是,通过重亚硫酸氢盐将未甲基化的胞嘧啶(c)转化为尿嘧啶(u),然后在pcr过程中,采用u耐受的聚合酶,将u识别为胸腺嘧啶(t),实现c到t的转化,分析时将测序数据分别比对到c到t和g到a转化的参考基因组,识别样本dna甲基化水平。在正常的人类dna中,约有3%至6%的c被甲基化,因此经过重亚硫酸氢盐转化的测序数据超过90%的c转化为t。基因组变异主要包含基因突变、拷贝数变异和结构变异。基因突变是指基因在结构上发生碱基对组成或排列顺序的改变,包括碱基替换、dna插入、dna缺失或dna重复引起的序列的改变。拷贝数变异一般是指长度为1kb到几个mb的基因组大片段的拷贝数复制、缺失。结构变异一般是指染色体重组,在基因组上距离很远的两个基因发生融合,形成了新的编码序列。基因突变、拷贝数变异、结构变异直接改变了dna碱基序列,影响生物的遗传特性,在肿瘤的早期诊断、指导用药及预后监测有重要作用。基因突变、拷贝数变异、结构变异都是dna分子核苷酸序列的改变,可以通过二代测序技术将测序数据与参考基因组进行比对分析检测得到。该检测方法产生的数据只包含dna碱基序列信息,无法识别碱基是否发生甲基化。插入片段通常是指在二代测序的文库构建中利用超声或者酶切技术将样本中的dna分子进行打断获得的dna片段。对于血液中游离dna(cfdna)片段,其长度分布在75~250bp之间,在文库构建前不需要将cfdna打断。通过插入片段分析结果可以反映插入片段的分布,例如cfdna片段的分布。目前,插入片段分析是通过基因组双端测序数据比对得出的,该检测数据不含有甲基化信息,无法进行甲基化相关分析。综上,目前基因组变异(基因突变、拷贝数变异、结构变异)分析和插入片段分析是基于dna测序数据与参考基因组比对进行差异分析,未考虑含甲基化标志信息的序列分析,无法进行含甲基化信号数据分析。目前的甲基化水平的检测是针对重亚硫酸氢盐转化测序(bisulfitesequencing,bs)数据与经过c到t和g到a转化的参考基因组的差异分析,进行所有位点甲基化水平的评估。由于bs测序数据中超过90%以上的c被转化,并且在转化过程会产生dna损伤,所以无法进行基因组变异和插入片段分析。因此,本领域目前仍不能实现对dna测序数据同时进行甲基化水平、基因组变异(基因突变、拷贝数变异、结构变异)和插入片段分析的检测。技术实现要素:本发明的目的在于实现对dna测序数据同时进行甲基化水平、基因组变异(基因突变、拷贝数变异、结构变异)和插入片段分析的检测。本发明通过以下技术方案来达到本发明的目的。在第一方面,本发明提供一种同时检测甲基化水平、基因组变异和插入片段的方法,其中基因组变异包括基因突变、拷贝数变异和结构变异,该方法包括以下步骤:s1:测序数据提供步骤,通过对包含待测样本dna与甲基化阳参dna和甲基化阴参dna的混合样本进行甲基化非重亚硫酸氢盐测序,提供测序数据;s2:测序数据处理步骤,将测序数据进行处理,得到甲基化阳参dna和甲基化阴参dna的有效数据及待测样本dna的有效数据;s3:条件判断步骤,根据甲基化阳参dna和甲基化阴参dna的有效数据,统计甲基化阳参dna在cpg区域的甲基化水平α和甲基化阴参dna整条基因组上的甲基化水平β,并判断α和β是否满足α≥95%且β<5%的条件,如果满足,则进行s4步骤,如果不满足,则返回s1步骤重新进行甲基化非重亚硫酸氢盐测序;s4:待测样本甲基化检测步骤,对待测样本dna的有效数据进行甲基化分析,并统计待测样本dna的甲基化水平;s5:待测样本基因突变检测步骤,对待测样本dna的有效数据进行基因突变分析,根据基因突变分析结果进行基因功能区域过滤和数据库频率过滤,得到第一突变集,根据甲基化统计结果去除甲基化转化的reads,并根据cpg、chg、chh设置向上浮动阈值进行第一突变集的过滤,得到最终突变集;s6:待测样本拷贝数变异检测步骤,根据待测样本dna的有效数据进行拷贝数变异分析,得到拷贝数变异数据,并进行过滤筛选;s7:待测样本结构变异检测步骤,根据待测样本dna的有效数据进行结构变异分析,得到结构变异数据,并进行过滤筛选;s8:待测样本插入序列检测步骤,根据待测样本dna的有效数据进行插入片段分析,将覆盖突变型和野生型的reads区分,分别统计突变型插入片段和野生型插入片段的分布结果。在本发明的具体实施方案中,该待测样本dna为人的体细胞dna,该甲基化阳参dna和甲基化阴参dna为与人类物种不同的物种的dna。在本发明的具体实施方案中,在s2步骤中,将测序数据进行处理包括将该测序数据进行接头去除、低质量序列过滤,其中低质量序列过滤条件为质量值<15的低质量碱基不超过该序列的50%;将过滤后的序列数据分别与人参考基因组、甲基化阳参dna和甲基化阴参dna进行比对,得到比对文件,并建立索引;合并多条lane得到的比对文件,并进行排序;合并后的比对文件去除pcr产生的重复序列,得到该有效数据。在本发明的具体实施方案中,在s4步骤中,样本甲基化分析结果包含所有c碱基甲基化信息,包括基因组位置信息、甲基化覆盖深度、非甲基化覆盖深度、甲基化频率;样本甲基化统计结果包含cpg二核苷和非cpg二核苷区域的甲基化水平,其中非cpg二核苷区域包括chh位点和chg位点,其中h为非g碱基。在本发明的具体实施方案中,在s5步骤中,样本基因突变分析结果包括基因组位点信息、突变信息,基因功能区域过滤为只保留外显子区域、错义突变、无义突变、移码突变,数据库频率过滤为去除千人基因组频率≥0.001,cpg的向上浮动阈值为0.1,chg和chh的向上浮动阈值为0.05。在本发明的具体实施方案中,在s6步骤中,该拷贝数变异数据过滤筛选条件为ratio>2或者ratio<0.5。在本发明的具体实施方案中,在s7步骤中,该结构变异数据筛选条件为断点覆盖度≥5。在本发明的具体实施方案中,在s8步骤中,突变型和野生型reads区分是根据bam文件的cigar和flag信息识别突变位点,其中突变位点的识别排除甲基化信号的影响。在第二方面,本发明提供一种用于实施本发明第一方面的同时检测甲基化水平、基因组变异和插入片段的方法的装置,其中基因组变异包括基因突变、拷贝数变异和结构变异,该装置包括以下模块:m1:测序数据提供模块,用于通过对包含待测样本dna与甲基化阳参dna和甲基化阴参dna的混合样本进行甲基化非重亚硫酸氢盐测序,提供测序数据;m2:测序数据处理模块,用于将测序数据进行处理,得到甲基化阳参dna和甲基化阴参dna的有效数据及待测样本dna的有效数据;m3:条件判断模块,用于根据甲基化阳参dna和甲基化阴参dna的有效数据,统计甲基化阳参dna在cpg区域的甲基化水平α和甲基化阴参dna整条基因组上的甲基化水平β,并判断α和β是否满足α≥95%且β<5%的条件,如果满足,则进行m4模块,如果不满足,则返回m1模块重新进行甲基化非重亚硫酸氢盐测序;m4:待测样本甲基化检测模块,用于对待测样本dna的有效数据进行甲基化分析,并统计待测样本dna的甲基化水平;m5:待测样本基因突变检测模块,用于对待测样本dna的有效数据进行基因突变分析,根据基因突变分析结果进行基因功能区域过滤和数据库频率过滤,得到第一突变集,根据甲基化统计结果去除甲基化转化的reads,并根据cpg、chg、chh设置向上浮动阈值进行第一突变集的过滤,得到最终突变集;m6:待测样本拷贝数变异检测模块,用于根据待测样本dna的有效数据进行拷贝数变异分析,得到拷贝数变异数据,并进行过滤筛选;m7:待测样本结构变异检测模块,用于根据待测样本dna的有效数据进行结构变异分析,得到结构变异数据,并进行过滤筛选;m8:待测样本插入序列检测模块,用于根据待测样本dna的有效数据进行插入片段分析,将覆盖突变型和野生型的reads区分,分别统计突变型插入片段和野生型插入片段的分布结果。在第三方面,本发明提供一种计算机可读介质,该计算机可读介质存储有计算机程序指令,其中当该计算机程序指令被处理器执行时,本发明第一方面的同时检测甲基化水平、基因组变异和插入片段的方法被运行。在第四方面,本发明提供一种用于实施本发明第一方面的同时检测甲基化水平、基因组变异和插入片段的方法的设备,该设备包括:用于存储计算机程序指令的存储器,和用于执行该计算机程序指令的处理器,其中当该计算机程序指令被该处理器执行时,该设备运行本发明第一方面的同时检测甲基化水平、基因组变异和插入片段的方法。本发明的有益效果:针对甲基化非重亚硫酸氢盐测序数据,实现从下机数据到甲基化水平、基因组变异(基因突变、拷贝数变异和结构变异)和插入片段多个维度的一步法检测分析。适用于甲基化非重亚硫酸氢盐的全基因组和靶向捕获数据类型,并可以进行单个癌症样本和成对样本(含对照样本的癌症样本)的分析。针对单个位点,可同时进行该位点甲基化和基因突变的检测,并给出甲基化频率和突变频率。基因突变筛选可去除甲基化信号影响和覆盖度不足引入的假阳性结果,过滤效率达99%。去除甲基化背景噪声影响,进行插入片段分析,可有效将突变型片段和野生型片段区分。甲基化非重亚硫酸氢盐测序的数据有效率和人类参考基因组比对率显著高于重亚硫酸氢盐转化测序(bs)的数据有效率和人类参考基因组比对率。附图说明图1显示本发明的同时检测甲基化水平、基因组变异和插入片段的方法的步骤流程图;图2显示本发明的同时检测甲基化水平、基因组变异和插入片段的装置的模块框图;图3显示本发明方法中的基因突变筛选可去除甲基化信号影响和覆盖度不足引入的假阳性结果,过滤效率达99.66%以上;图4显示显示本发明方法去除甲基化背景噪音影响,进行插入片段分析,可有效将突变型片段和野生型片段区分;图5显示本发明方法使用的甲基化非重亚硫酸氢盐测序法与全基因组甲基化重亚硫酸氢盐测序法的比较。具体实施方式为了使本发明所解决的技术问题、所采用的技术方案及所获得的有益效果更加清楚明白,以下结合附图及具体实施例对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。本领域目前仍不能实现对dna测序数据同时进行甲基化水平、基因组变异(基因突变、拷贝数分析、结构变异)和插入片段分析的检测。简单地说,现有的基因组甲基化检测方法通常是重亚硫酸氢盐转化测序法(bisulfitesequencing,bs),将bs数据与经过c到t和g到a转化的参考基因组进行差异分析来进行所有位点甲基化水平的评估。由于bs测序数据中超过90%以上的c被转化,并且在转化过程会产生dna损伤,所以无法进行基因组变异和插入片段分析。通过二代测序技术将测序数据与参考基因组进行比对分析,可以检测基因突变、拷贝数变异、结构变异,但该检测方法产生的数据只包含dna碱基序列信息,无法识别碱基是否发生甲基化。再且,目前插入片段分析是通过基因组双端测序数据比对得出的,该检测数据不含有甲基化信息,无法进行甲基化相关分析。甲基化非重亚硫酸氢盐测序(英文为oxidation-reductionmethylatedc-assistedsequenceing,简称omas)技术是一种新的甲基化检测技术,其涉及在tet酶辅助下将dna基因组的甲基化c碱基转化为5-醛基胞嘧啶(5fc)和5-羧基胞嘧啶(5cac),然后经过硼烷还原为t碱基。该技术仅将5-甲基胞嘧啶(5mc)和5-羟甲基胞嘧啶(5mc)等修饰c碱基(约占胞嘧啶总量的4~5%)最终转化为t碱基。由该检测技术的测序数据可进行每个位点甲基化水平的检测及甲基化水平评估。有关甲基化非重亚硫酸氢盐测序技术的操作,可例如参见本发明申请人的申请号为cn201911159400、发明名称为“全基因组甲基化非重亚硫酸氢盐测序文库及构建”的中国专利申请,该申请的公开内容以引用方式全部并入本文。本发明人研究发现,甲基化非重亚硫酸氢盐测序技术对基因组改变微小,可以同时进行基因组变异和插入片段分析。但是,目前还没有针对甲基化非重亚硫酸氢盐测序数据同时进行甲基化水平、基因组变异(基因突变、拷贝数分析、结构变异)和插入片段分析的检测的报道。因此,本发明人开创性地提出了一种利用甲基化非重亚硫酸氢盐测序技术同时检测甲基化水平、基因组变异和插入片段的方法,其中基因组变异包括基因突变、拷贝数变异和结构变异。该方法的步骤流程图如图1所示,以下对该方法的各个步骤进行详细的说明。s1:测序数据提供步骤,通过对包含待测样本dna与甲基化阳参dna和甲基化阴参dna的混合样本进行甲基化非重亚硫酸氢盐测序,提供测序数据。具体地讲,待测样本dna为人的体细胞dna,包括但不限于人新鲜组织来源dna、石蜡包埋组织来源dna、血浆来源dna、胸水来源dna、腹水来源dna。甲基化阳参dna和甲基化阴参dna为与人类物种不同的物种的dna。甲基化阳参可例如直接采用甲基化的puc19(zymoresearch),也可例如采用未甲基化puc19通过m.ssi甲基化转移酶合成的甲基化puc19;甲基化阴参dna可例如直接采用lambdadna(promega)。甲基化非重亚硫酸氢盐测序数据可以为全基因组数据和靶向捕获数据,其中靶向捕获数据包括全外显子数据。测序数据可以为单个测试样本(例如癌症样本)的测序数据,也可以为含有对照样本的测试样本(例如癌症样本)的测序数据,其中对照样本例如为受试者本人的外周血分离的白细胞或者癌旁样本。s2:测序数据处理步骤,将测序数据进行处理,得到甲基化阳参dna和甲基化阴参dna的有效数据及待测样本dna的有效数据。具体地讲,将测序数据进行处理包括将测序数据进行接头去除、低质量序列过滤,其中低质量序列过滤条件为质量值<15的低质量碱基不超过该序列的50%;将过滤后的序列数据分别与人参考基因组、甲基化阳参dna和甲基化阴参dna进行比对,得到比对文件,并建立索引;合并多条lane得到的比对文件,并进行排序;合并后的比对文件去除甲基化非重亚硫酸氢盐测序中进行的pcr所产生的重复序列,得到有效数据。可以利用fastp或者trimgalore软件进行接头去除、低质量序列过滤,过滤后数据格式为fastq。可以使用bwa软件将过滤后的数据与甲基化阳参dna、甲基化阴参dna和人参考基因组进行比对,得到bam格式的比对文件。建立索引可以通过samtools(index)软件建立后缀为.bai的索引文件。合并多条lane数据可以通过samtools(merge)软件进行合并。去除pcr产生的重复序列,可以通过gatk调用picard包进行重复序列的去除。s3:条件判断步骤,根据甲基化阳参dna和甲基化阴参dna的有效数据,统计甲基化阳参dna在cpg区域的甲基化水平α和甲基化阴参dna整条基因组上的甲基化水平β,并判断α和β是否满足α≥95%且β<5%的条件,如果满足,则进行s4步骤,如果不满足,则返回s1步骤重新进行甲基化非重亚硫酸氢盐测序。具体地讲,甲基化阳参和甲基化阴参的甲基化水平是比对文件cpg二核苷位点碱基c到t转化reads占比的平均值。可以通过astaircall统计阳参在cpg区域的甲基化水平α和阴参整条基因组上的甲基化水平β。不满足α≥95%且β<5%的条件,则说明甲基化非重亚硫酸氢盐转化不成功,需重新进行甲基化非重亚硫酸氢盐转化,即返回s1步骤重新进行甲基化非重亚硫酸氢盐测序。s4:待测样本甲基化检测步骤,对待测样本dna的有效数据进行甲基化分析,并统计待测样本dna的甲基化水平。具体地讲,样本甲基化分析结果包含所有c碱基甲基化信息,包括基因组位置信息、甲基化覆盖深度、非甲基化覆盖深度、甲基化频率;样本甲基化统计结果包含cpg二核苷和非cpg二核苷区域的甲基化水平,其中非cpg二核苷区域包括chh位点和chg位点,其中h为非g碱基。统计样本的甲基化水平的方法与步骤s3中统计甲基化阴参的甲基化水平的方法一致。s5:待测样本基因突变检测步骤,对待测样本dna的有效数据进行基因突变分析,根据基因突变分析结果进行基因功能区域过滤和数据库频率过滤,得到第一突变集,根据甲基化统计结果去除甲基化转化的reads,并根据cpg、chg、chh设置向上浮动阈值进行第一突变集的过滤,得到最终突变集。具体地讲,样本基因突变检测可以使用突变检测软件bcftools、mutect2、varscan进行。样本基因突变分析结果包括基因组位点信息、突变信息,其中基因组位点信息包括染色体号和突变起始位值,突变信息包括野生型、突变型及其reads覆盖数目、突变频率、突变注释信息(包含基因、功能、数据库注释信息)。基因功能区域过滤为只保留外显子区域、错义突变、无义突变、移码突变,数据库频率过滤为去除千人基因组频率≥0.001。cpg的向上浮动阈值为0.1,chg和chh的向上浮动阈值为0.05。即,对于cpg位点,去除c到t或者g到a突变的频率与该位点甲基化频率之差<0.1的突变,对于其它c位点,去除c到t或者g到a突变的频率与该位点甲基化频率之差<0.05的突变。突变注释可以使用软件annovar、vep来进行。s6:待测样本拷贝数变异检测步骤,根据待测样本dna的有效数据进行拷贝数变异分析,得到拷贝数变异数据,并进行过滤筛选。具体地讲,拷贝数变异分析可以利用freec、cnvnator软件来进行,拷贝数变异数据包括基因区域信息和该区域包含的基因信息,拷贝数变异数据过滤筛选条件为ratio>2或者ratio<0.5。s7:待测样本结构变异检测步骤,根据待测样本dna的有效数据进行结构变异分析,得到结构变异数据,并进行过滤筛选。具体地讲,结构变异分析可以利用manta软件来进行,结构变异数据筛选条件为断点覆盖度≥5。s8:待测样本插入序列检测步骤,根据待测样本dna的有效数据进行插入片段分析,将覆盖突变型和野生型的reads区分,分别统计突变型插入片段和野生型插入片段的分布结果。具体地讲,突变型和野生型reads区分是根据bam文件的cigar和flag信息识别突变位点,其中突变位点的识别排除甲基化信号的影响。本发明人在提出了一种利用甲基化非重亚硫酸氢盐测序技术同时检测甲基化水平、基因组变异和插入片段的方法的基础上,相应地提出了一种用于实施该方法的装置,该装置的模块框图如图2所示,该装置包括以下模块:m1:测序数据提供模块,用于通过对包含待测样本dna与甲基化阳参dna和甲基化阴参dna的混合样本进行甲基化非重亚硫酸氢盐测序,提供测序数据;m2:测序数据处理模块,用于将测序数据进行处理,得到甲基化阳参dna和甲基化阴参dna的有效数据及待测样本dna的有效数据;m3:条件判断模块,用于根据甲基化阳参dna和甲基化阴参dna的有效数据,统计甲基化阳参dna在cpg区域的甲基化水平α和甲基化阴参dna整条基因组上的甲基化水平β,并判断α和β是否满足α≥95%且β<5%的条件,如果满足,则进行m4模块,如果不满足,则返回m1模块重新进行甲基化非重亚硫酸氢盐测序;m4:待测样本甲基化检测模块,用于对待测样本dna的有效数据进行甲基化分析,并统计待测样本dna的甲基化水平;m5:待测样本基因突变检测模块,用于对待测样本dna的有效数据进行基因突变分析,根据基因突变分析结果进行基因功能区域过滤和数据库频率过滤,得到第一突变集,根据甲基化统计结果去除甲基化转化的reads,并根据cpg、chg、chh设置向上浮动阈值进行第一突变集的过滤,得到最终突变集;m6:待测样本拷贝数变异检测模块,用于根据待测样本dna的有效数据进行拷贝数变异分析,得到拷贝数变异数据,并进行过滤筛选;m7:待测样本结构变异检测模块,用于根据待测样本dna的有效数据进行结构变异分析,得到结构变异数据,并进行过滤筛选;m8:待测样本插入序列检测模块,用于根据待测样本dna的有效数据进行插入片段分析,将覆盖突变型和野生型的reads区分,分别统计突变型插入片段和野生型插入片段的分布结果。进一步地,本发明人提出了一种计算机可读介质,该计算机可读介质存储有计算机程序指令,其中当该计算机程序指令被处理器执行时,本发明第一方面的同时检测甲基化水平、基因组变异和插入片段的方法被运行。还进一步地,本发明人提出了一种用于实施本发明第一方面的同时检测甲基化水平、基因组变异和插入片段的方法的设备,该设备包括:用于存储计算机程序指令的存储器,和用于执行该计算机程序指令的处理器,其中当该计算机程序指令被该处理器执行时,该设备运行本发明第一方面的同时检测甲基化水平、基因组变异和插入片段的方法。以下通过实施例和比较例对本发明作进一步的举例说明。实施例1:本发明方法的应用实例s1步骤:取100ng人血液cfdna、0.2ng甲基化puc19dna(甲基化阳参dna)、未甲基化的lambdadna(甲基化阴参dna)进行混合进行打断,通过甲基化非重亚硫酸氢盐测序。测序文库的构建参照本发明申请人的申请号为cn201911159400、发明名称为“全基因组甲基化非重亚硫酸氢盐测序文库及构建”的中国专利申请的实施例4,测序平台采用gene+seq平台。测序后,得到下机数据l1_r1.fq.gz、l1_r2.fq.gz、l1_r1.clean.fq.gz、l2_r2.clean.fq.gz。s2步骤:通过命令fastp-ir1.fq.gz-ir2.fq.gz-or1.clean.fq.gz-or2.clean.fq.gz将下机数据进行接头去除、质量过滤,得到过滤后的序列数据,如下面表格所示:然后通过命令bwamem-otest.bam-mhas.fal1_r1.clean.fq.gal1_r2.clean.fq.gz将过滤后的序列数据分别与阳参、阴参、人参考基因组进行比对,得到比对文件(bam格式),并利用samtoolsindex命令建立索引。分别使用命令samtoolsmerge和samtoolssort合并多条lane的比对文件,对合并后的文件排序,并统计比对结果,如下面表格所示:然后,利用java-xmx20g-djava.io.tmpdir=./-jarpicard.jarmarkduplicatesi=test.bamo=test.mark.bamm=a.metrics去除pcr产生的重复序列,并统计结果,如下面表格所示:s3步骤:通过astaircall统计甲基化阳参在cpg区域的甲基化水平α和甲基化阴参整条基因组上的甲基化水平β。结果满足α≥95%且β<5%的条件,如下面表格所示:sampleαβ2003270172pd95.31%0.67%s4步骤:通过astaircall检测样本甲基化水平,统计结果(部分)如下面表格所示:然后,根据样本甲基化检测结果,统计基因组不同区域的甲基化水平,如下面表格所示:samplecpgchgchhc2003270172pd64.32%0.35%0.31%2.58%s5步骤:根据待测样本dna的有效数据采用samtools+bcftools进行样本基因突变分析,进行基因功能区域过滤和数据库频率过滤,得到第一突变结果,然后进行甲基化噪音过滤,得到最终突变结果,如下面表格所示:s6步骤:调用命令freec-confconfig_wgs.txt进行拷贝数变异分析,并进行过滤筛选,得到最终拷贝数分析结果,如下面表格所示:samplegenecnvtype2003270172pderbb24.054828gains7步骤:调用命令configmanta进行结构变异检测及筛选。s8步骤:自写代码进行插入片段分析,根据bam文件提取插入片段信息(第9列),根据cigar和flag信息,区分突变型和野生型的reads,并分别输出两个文件(1个为突变型reads的插入片段;1个为野生型reads的插入片段),然后绘制插入片段的密度分布图(将突变型和野生型reads的插入片段分布放在一张图中)。上述s1步骤至s8步骤,可以通过调用软件multi_analyse,一步法得到分析结果,具体命令如下:perlmulti_analyse\--ref_puc19puc19.fa\--ref_lambdalambda.fasta\--refhs37d5.fa\--bedhs37d5.region.bed\--chrfilechromosome/\sample.list其中sample.list为:machineno:geneseq2000samplenoidnosampletypecasecase2003270172pdp2003270172pd_hum_c_gc0c_4014_z_0_a,2020-04-19运行结果:甲基化检测结果:typemethylationlevelmethylationdepthalldepthcpg69.78%2819785440407938chg0.44%10161172.3e+08chh0.40%32943788.24e+08基因突变检测结果(部分):chrposrefaltinfo116893254ag0.16|(12,14,3,2)11.21e+08tc0.24|(38,51,9,19)11.43e+08ac0.28|(19,9,8,3)进一步地,图3显示本发明方法中的基因突变筛选可去除甲基化信号影响和覆盖度不足引入的假阳性结果,过滤效率达99.66%以上。拷贝数变异检测结果(部分):chrstartendcnv221624500016344000loss221634400016353000gain221635300016851000loss221685100017037000gain结构变异检测结果(部分):chrom_astart_aend_achrom_bstart_bend_btype2790765697907657227908120579081208del21793010451793010462179306335179306336del32034297920342980hs37d591064079106408bnd3800644698006447538006510680065107bnd3800651018006510738006447480064475bnd380325189803251906121481643121481644bnd41159287181159287234115931872115931877del612148164312148164438032518980325190bnd插入片段检测结果(部分):注:mutgroup为覆盖突变位点的插入片段组;wildgroup为覆盖非突变位点的插入片段组。进一步参见图4,其中显示了本发明方法去除甲基化背景噪音影响,进行插入片段分析,可有效将突变型片段和野生型片段区分。比较例1:甲基化非重亚硫酸氢盐测序法和全基因组甲基化重亚硫酸氢盐测序法的比较取100ng人血液cfdna,分别进行甲基化非重亚硫酸氢盐测序(omas)和全基因组甲基化重亚硫酸氢盐测序(wgbs),并进行数据有效率(effective_rate)和人参考基因组比对率(mappingrate)的比较。甲基化非重亚硫酸氢盐测序参照实施例1的s1步骤进行;全基因组甲基化重亚硫酸氢盐测序(wgbs)参照中国专利文献cn104532360b中公开的全基因组甲基化测序文库及其构建方法构建,其中“c”到“t”的转化处理采用重亚硫酸氢盐转化进行;测序平台采用illumina(hiseq)。结果在图5显示,可见与wgbs相比,本发明方法采用了omas,数据有效率和人参考基因组比对率分别为约93%和95%,显著高于wgbs的约84%的数据有效率和约85%的人参考基因组比对率。这是因为wgbs将基因组中未甲基化的c转为t,然后建库测序。由于碱基不平衡导致测序数据碱基质量低,因此数据有效率低,并且在人参考基因组比对分析中,通常使用bismarker软件将数据分别比对到经过c到t转化的人基因组和经过g到a转化的人基因组,比对率也较低。本对比例说明,由于c到t转化的原理不同,omas的数据有效率和人参考基因组比对率显著高于wgbs的数据有效率和人参考基因组比对率。因此,本发明方法由于采用了omas,不仅在碱基转化过程中大大减少对dna的损伤,使得可以实现同时检测甲基化水平、基因组变异和插入片段,并且可以提高数据有效率和人参考基因组比对率,提高检测的准确度。以上应用了具体实例对本发明进行了阐述,只是用于帮助理解本发明,并不用以限制本发明。本发明所属
技术领域:
的技术人员依据本发明的构思,还可以做出若干简单推演、变形或替换。这些推演、变形或替换方案也落入本发明的权利要求范围内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1