一种基于二次过滤的遗传代谢病特异性指标挖掘方法与流程
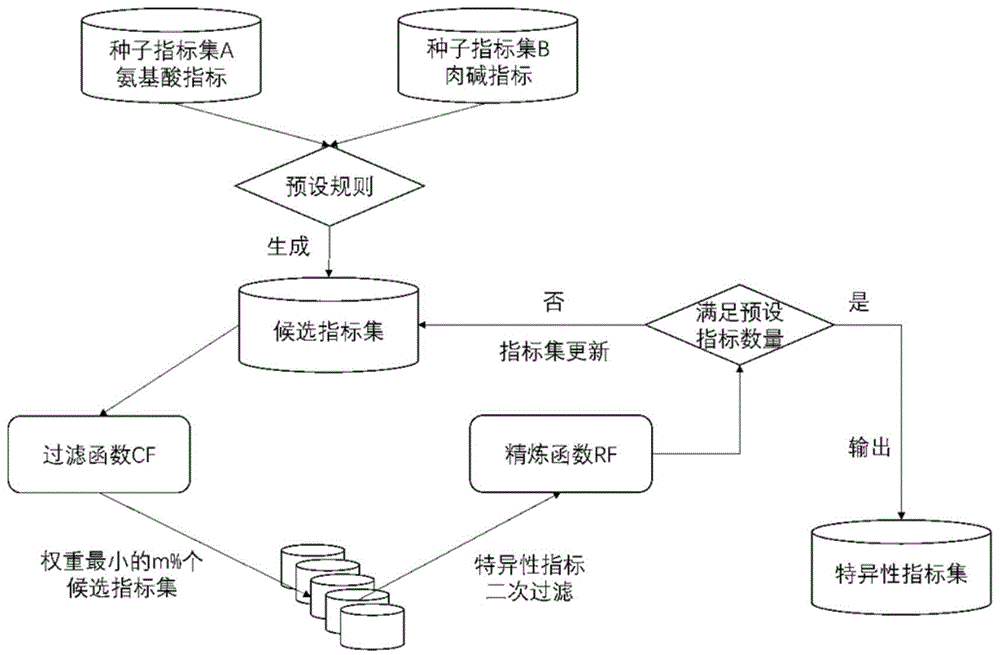
本发明属于信息技术领域,具体涉及一种基于二次过滤的遗传代谢病特异性指标挖掘方法。
背景技术:
随着生物技术的发展,串联质谱法逐渐成为遗传代谢病的主要筛查方法。在一次实验室检验中,串联质谱法可以同时测定几十种遗传代谢病相关代谢物的血液浓度(筛查指标),对数十种遗传代谢病做出同步诊断。但在现有的筛查系统中,对于某一种遗传代谢病所使用的特异性指标一般不会超过五个,导致筛查指标使用效率低下,多样性不足,间接影响了筛查的准确度。
现阶段,遗传代谢病特异性指标的选取主要来自业界共识、相关医学文献和医生临床经验。这些方法往往依靠医学专家对指标进行人工分析,选取单个指标或指标比值,难以同时考虑几十种筛查指标之间、与疾病之间复杂的关联关系。相对地,随着机器学习和统计理论的广泛应用,数据驱动的特异性指标挖掘方法能够自动高效地发现筛查指标的关联,解决以往依靠人工寻找指标的问题。然而,现有方法还存在类型单一(基于模型、基于统计、基于搜索其中一种)和计算过程单一(一次性计算,没有迭代精炼结果)的问题。
为此,本发明结合统计理论和机器学习模型,提出了一种多次迭代过滤的特异性指标挖掘方法。首先,本发明通过一系列预设规则来构建候选指标集,使可供搜索的筛查指标更为丰富;其次,本发明提出了基于候选指标集合的二次筛选方法,使筛选得到的筛查指标关联性更强;最后,通过不断迭代筛选,得到一系列特异性强的筛查指标,在一定程度上提高整体筛查的准确度。
技术实现要素:
目前遗传代谢病的筛查指标主要依靠人工分析和选取,难以同时考虑几十种指标之间的复杂关系,导致现有特异性筛查指标存在数量少、多样性不足、使用率低的问题。本发明提供了一种基于二次过滤的遗传代谢病特异性指标挖掘方法,利用预设规则构建候选指标集,设计过滤函数和精练函数评估候选指标集之间、候选指标集内部的特异性程度,通过不断迭代自动挖掘出遗传代谢病相关的特异性指标,提高筛查指标的多样性和筛查的准确度。
一种基于二次过滤的遗传代谢病特异性指标挖掘方法,包含以下步骤:
1.候选指标集构建
1)根据临床筛查指标和筛查对象的信息,构建两组种子指标集:a,b;其中a表示氨基酸指标,b表示肉碱指标;n(a)表示a包含的指标个数,n(b)表示b包含的指标个数;
2)对于种子指标集a中的任意一个种子指标ai,其中1≤i≤n(a),构建ai所属的候选指标集cand(ai);其中cand(ai)的构建方法为:(1)将ai、ai的平方、ai的对数加入cand(ai);(2)将ai+aj、ai/aj加入cand(ai),其中aj为a中除了ai以外的n(a)-1个种子指标;(3)将(ai+aj)/ak、ak/(ai+aj)加入cand(ai),其中aj为a中除了ai以外的n(a)-1个种子指标,ak为a中除了ai和aj以外的n(a)-2个种子指标;
3)对于种子指标集b中的任意一个种子指标bi,其中1≤i≤n(b),构建bi所属的候选指标集cand(bi);其中cand(bi)的构建方法为:(1)将bi、bi的平方、bi的对数加入cand(bi);(2)将bi+bj、bi/bj加入cand(bi),其中bj为b中除了bi以外的n(b)-1个种子指标;(3)将(bi+bj)/bk、bk/(bi+bj)加入cand(bi),其中bj为b中除了bi以外的n(b)-1个种子指标,bk为b中除了bi和bj以外的n(b)-2个种子指标;
4)最终一共获得n(a)+n(b)组候选指标集,共7*(n(a)+n(b))个候选指标,包括cand(ai)和cand(bj),其中1≤i≤n(a),1≤j≤n(b)。
2.候选指标集过滤
1)选取一个基于线性空间的机器学习算法(如逻辑回归算法),设定待优化的目标函数,使用构建的所有候选指标集进行训练,其中设定待优化的目标函数为算法原目标函数加上候选指标集过滤函数cf:
其中wi和wj为候选指标集cand(ai)和cand(bj)对应的权重,‖·‖2为二范数,n(·)为集合内元素的个数。
3.候选指标集精炼
1)对于步骤2得到的所有权重值‖w*‖2,选取前m%个(如前10%个)权重值最小的候选指标集;
2)对于选取的m%个候选指标集中的所有指标,根据精练函数rf计算每个指标的特异性:
其中x为候选指标集内的一个指标,y为一种遗传代谢病的诊断结论,‖wx‖2为x所在候选指标集的权重值,pr(x)和pr(y)为边缘概率,pr(x,y)为联合概率;
3)将特异性最小的n%个(如20%个)指标从所在候选指标集中剔除。
4.特异性指标迭代挖掘
重复迭代步骤2与步骤3,直至剩余指标数量为步骤1构建得到的候选指标的s%(如5%)。
其中,氨基酸指标和肉碱指标具体如下:
本发明的有益效果为:
本发明的基于二次过滤的遗传代谢病特异性指标挖掘方法,首先通过一系列预设规则来构建候选指标集,使可供搜索的筛查指标更为丰富;
其次提出了基于候选指标集合的二次筛选方法,使筛选得到的筛查指标关联性更强,利用过滤函数分组评估候选指标集的特异性,利用精练函数保留候选指标集中特异性更高的筛查指标;
最后,利用特异性指标挖掘方法自动分析筛查指标间的复杂关系,搜索到最具有特异性的筛查指标,无需人工干预;
利用本发明方法挖掘得到的特异性指标能够将现有遗传代谢病筛查的初筛假阳性率从3%降低至1%左右。
附图说明
图1为本发明的基于二次过滤的遗传代谢病特异性指标挖掘方法。
具体实施方式
下面结合附图和具体实施例对本发明进行进一步地说明。
如图1为本发明的基于二次过滤的遗传代谢病特异性指标挖掘方法,包括如下步骤:
1.候选指标集构建
1)根据临床筛查指标和筛查对象的信息,构建两组种子指标集:a,b;其中a表示氨基酸指标,b表示肉碱指标;n(a)表示a包含的指标个数,n(b)表示b包含的指标个数;
2)对于种子指标集a中的任意一个种子指标ai,其中1≤i≤n(a),构建ai所属的候选指标集cand(ai);其中cand(ai)的构建方法为:(1)将ai、ai的平方、ai的对数加入cand(ai);(2)将ai+aj、ai/aj加入cand(ai),其中aj为a中除了ai以外的n(a)-1个种子指标;(3)将(ai+aj)/ak、ak/(ai+aj)加入cand(ai),其中aj为a中除了ai以外的n(a)-1个种子指标,ak为a中除了ai和aj以外的n(a)-2个种子指标;
3)对于种子指标集b中的任意一个种子指标bi,其中1≤i≤n(b),构建bi所属的候选指标集cand(bi);其中cand(bi)的构建方法为:(1)将bi、bi的平方、bi的对数加入cand(bi);(2)将bi+bj、bi/bj加入cand(bi),其中bj为b中除了bi以外的n(b)-1个种子指标;(3)将(bi+bj)/bk、bk/(bi+bj)加入cand(bi),其中bj为b中除了bi以外的n(b)-1个种子指标,bk为b中除了bi和bj以外的n(b)-2个种子指标;
4)最终一共获得n(a)+n(b)组候选指标集,共7*(n(a)+n(b))个候选指标,包括cand(ai)和cand(bj),其中1≤i≤n(a),1≤j≤n(b)。
2.候选指标集过滤
1)选取一个基于线性空间的机器学习算法(如逻辑回归算法),设定待优化的目标函数,使用构建的所有候选指标集进行训练,其中设定待优化的目标函数为算法原目标函数加上候选指标集过滤函数cf:
其中wi和wj为候选指标集cand(ai)和cand(bj)对应的权重,‖·‖2为二范数,n(·)为集合内元素的个数。
3.候选指标集精炼
1)对于步骤2得到的所有权重值‖w*‖2,选取前m%个(如前10%个)权重值最小的候选指标集;
2)对于选取的m%个候选指标集中的所有指标,根据精练函数rf计算每个指标的特异性:
其中x为候选指标集内的一个指标,y为一种遗传代谢病的诊断结论,‖wx‖2为x所在候选指标集的权重值,pr(x)和pr(y)为边缘概率,pr(x,y)为联合概率;
3)将特异性最小的n%个(如20%个)指标从所在候选指标集中剔除。
4.特异性指标迭代挖掘
重复迭代步骤2与步骤3,直至剩余指标数量为步骤1构建得到的候选指标的s%(如5%)。
其中,氨基酸指标和肉碱指标具体如下:
- 还没有人留言评论。精彩留言会获得点赞!