一种智能化药物靶点亲和力预测方法与流程与流程
本发明涉及药物靶点亲和力分析预测技术领域,尤其涉及一种智能化药物靶点亲和力预测方法与流程。
背景技术:
药物靶点是指药物在体内的作用结合位点,包括基因位点、受体、酶、离子通道、核酸等生物大分子。现代新药研究与开发的关键首先是寻找、确定和制备药物筛选靶—分子药靶。药物靶点是指药物在体内的作用结合位点,包括基因位点、受体、酶、离子通道、核酸等生物大分子。选择确定新颖的有效药靶是新药开发的首要任务。迄今已发现作为治疗药物靶点的总数约500个,其中受体尤其是g-蛋白偶联的受体(gpcr)靶点占绝大多数,另还有酶、抗菌、抗病毒、抗寄生虫药的作用靶点。
药物靶点亲和力预测方法与流程就是一种对药物与靶点之间的作用关系进行分析预测药物与靶点之间的亲和力,在对药物与靶点之间相互作用关系分析过程中,蛋白质序列特征向量存在许多冗余信息,特征维度搞,影响分类预测的准确率和效率,同时在进行分析缺少对药物分子化学结构进行分析,导致预测的结果准确性差。
技术实现要素:
本发明的目的在于提供一种智能化药物靶点亲和力预测方法与流程,实现对蛋白质序列特征向量存在许多冗余信息的去除,降低特征维度,提供预测的准确率和效率,同时通过数理统计方法建立药物化学机构与靶点的定量构效关系,实现计算药物化学结构亲和力预测,实现对药物与靶点亲和力进行双重方式预测分析,可以对预测的结果进行整合分析,显著提高预测的准确性。
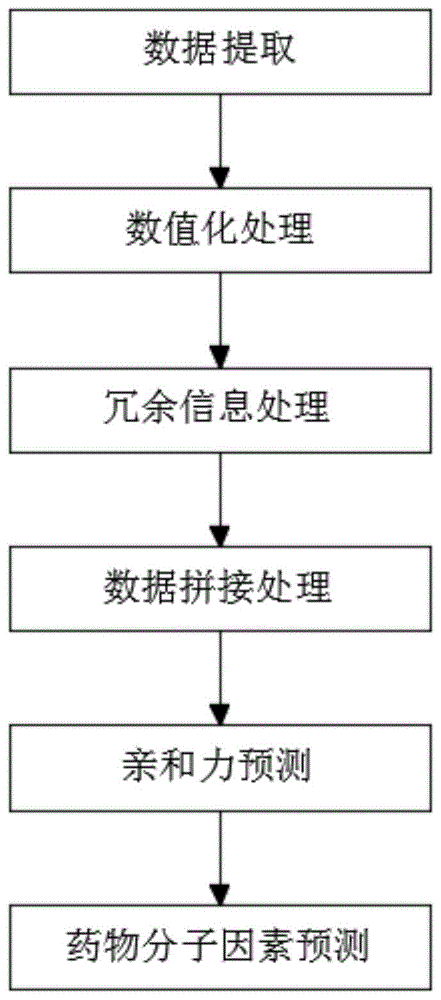
为了实现上述目的,本发明采用了如下技术方案:一种智能化药物靶点亲和力预测方法与流程,包括以下步骤:
s1:数据提取,从生物信息公共数据库中收集药物化合物分子数据和蛋白质序列;
s2:数值化处理,分别对药物化合物分子和蛋白质氨基酸序列进行数值化处理,获取药物化合物分子指纹特征向量x和蛋白质序列特征向量y;
s3:冗余信息处理,去除蛋白质序列特征向量y冗余信息,提取用于训练、学习和分类的高级特征,降低特征维度,提高药物与靶点相互预制预测的准确率;
s4:数据拼接处理,将蛋白质序列特征向量y和药物化合物分子指纹特征向量x进行拼接,获取药物靶点融合特征数据y;
s5:亲和力预测,根据获取的融合特征数据y,通过深度学习卷积神经网络分类器对药物靶点相互作用进行分析,实现对药物靶点亲和力预测;
s6:药物分子因素预测,获取药物化合物分子化学结构特征,通过数理统计方法建立药物化学机构与靶点的定量构效关系,实现计算药物化学结构亲和力预测。
作为上述技术方案的进一步描述:
所述步骤s2中药物化合物分子和蛋白质氨基酸序列进行数值化处理的使用的方法分别为分子指纹特征的药物化合物分子表征方法和得分特异性矩阵的蛋白质氨基酸序列表征方法。
作为上述技术方案的进一步描述:
所述步骤s3去除蛋白质序列特征向量y冗余信息具体方法为采用变分自动编码器提取蛋白质序列高级特征,降低维度,去除冗余信息。
作为上述技术方案的进一步描述:
所述步骤s4中数据拼接的规则为药物化合物分子指纹特征向量x在前,蛋白质序列特征向量y在后。
作为上述技术方案的进一步描述:
所述步骤s4中数据拼接处理具体方法为,通过串联规则将药物化合物药物化合物分子指纹特征向量x(x1、x2...xn)和蛋白质序列特征向量y(y1、y2...yn)进行同一规则拼接,获得药物靶点融合特征数据z(x1、x2...xn,y1、y2...yn)。
作为上述技术方案的进一步描述:
所述步骤s5亲和力预测具体方法为将融合特征数据y作为卷积神经网络的输入,对融合特征数据y进行训练生成分类器模型,利用反向传播、梯度下降方法调整分类器网络权值,然后获取最终亲和力预测结果。
作为上述技术方案的进一步描述:
所述步骤s6中药物化合物分子特征包括电性参数、物化参数和构型参数。
本发明提供了一种智能化药物靶点亲和力预测方法与流程。具备以下有益效果:
该智能化药物靶点亲和力预测方法与流程通过变分自动编码器实现对蛋白质序列特征向量存在许多冗余信息的去除,降低特征维度,提高预测的准确率和效率,同时通过数理统计方法建立药物化学机构与靶点的定量构效关系,实现计算药物化学结构亲和力预测,实现对药物与靶点亲和力进行双重方式预测分析,可以对预测的结果进行整合分析,显著提高预测的准确性。
附图说明
图1为本发明提出的一种智能化药物靶点亲和力预测方法与流程示意图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。
参照图1,一种智能化药物靶点亲和力预测方法与流程,包括以下步骤:
s1:数据提取,从生物信息公共数据库中收集药物化合物分子数据和蛋白质序列;
s2:数值化处理,分别对药物化合物分子和蛋白质氨基酸序列进行数值化处理,获取药物化合物分子指纹特征向量x和蛋白质序列特征向量y;
s3:冗余信息处理,去除蛋白质序列特征向量y冗余信息,提取用于训练、学习和分类的高级特征,降低特征维度,提高药物与靶点相互预制预测的准确率;
s4:数据拼接处理,将蛋白质序列特征向量y和药物化合物分子指纹特征向量x进行拼接,获取药物靶点融合特征数据y;
s5:亲和力预测,根据获取的融合特征数据y,通过深度学习卷积神经网络分类器对药物靶点相互作用进行分析,实现对药物靶点亲和力预测;
s6:药物分子因素预测,获取药物化合物分子化学结构特征,通过数理统计方法建立药物化学机构与靶点的定量构效关系,实现计算药物化学结构亲和力预测。
步骤s2中药物化合物分子和蛋白质氨基酸序列进行数值化处理的使用的方法分别为分子指纹特征的药物化合物分子表征方法和得分特异性矩阵的蛋白质氨基酸序列表征方法。
药物化合物分子和蛋白质氨基酸序列都是以一种复杂的字符编码保存在生物的数据库中,计算机智能算法难以运算,通过分子指纹特征的药物化合物分子表征方法和得分特异性矩阵的蛋白质氨基酸序列表征方法有效的将药物分子与蛋白质序列进行数值化表征,提取出药物化合物分子与靶标蛋白氨基酸序列的生物信息和本质属性,为实验运算提供数据保障。
步骤s3去除蛋白质序列特征向量y冗余信息具体方法为采用变分自动编码器提取蛋白质序列高级特征,降低维度,去除冗余信息。
蛋白质序列特征向量y存在许多冗余信息,特征维度高,影响预测的准确率与效率,通过变分自动编码器有效提取用于训练、学习、分类的高级特征,尽可能减少特征的维度,提高预测的准确率。
步骤s4中数据拼接的规则为药物化合物分子指纹特征向量x在前,蛋白质序列特征向量y在后。
步骤s4中数据拼接处理具体方法为,通过串联规则将药物化合物药物化合物分子指纹特征向量x(x1、x2...xn)和蛋白质序列特征向量y(y1、y2...yn)进行同一规则拼接,获得药物靶点融合特征数据z(x1、x2...xn,y1、y2...yn)。
由于药物化合物分子指纹特征向量x和蛋白质序列特征向量y数据信息样本众多,数据维度大,传统的分类预测方法难以达到很好的分类效果与预测速度,串联规则实现数据拼接处理提高药物靶点相互作用亲和力预测准确性和预测效率。
步骤s5亲和力预测具体方法为将融合特征数据y作为卷积神经网络的输入,对融合特征数据y进行训练生成分类器模型,利用反向传播、梯度下降方法调整分类器网络权值,然后获取最终亲和力预测结果。
步骤s6中药物化合物分子特征包括电性参数、物化参数和构型参数。
通过数理统计方法建立药物化学机构与靶点的定量构效关系,实现计算药物化学结构亲和力预测,实现对药物与靶点亲和力进行双重方式预测分析,可以对预测的结果进行整合分析,显著提高预测的准确性。
在本说明书的描述中,参考术语“一个实施例”、“示例”、“具体示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料过着特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!