一种通过机器学习算法预测化学品致突变性的方法与流程
1.本发明涉及一种通过建立qsar模型预测化学品致突变性的方法,属于生态风险评价测试策略领域。
背景技术:
2.致突变性指的是诱导遗传性损伤的能力。遗传物质发生改变可以通过以下几种方式:基因突变;染色体畸变;染色体数目改变。基因突变指的是基因组dna分子发生的突然地、可遗传的变异现象。其中包括碱基对的置换以及碱基对的增加或减少。染色体畸变则是指染色体在结构上发生变化。所以能够致使突变发生的诱变剂也就分为两类。一类是直接作用于dna的诱变剂,另一类是作用于染色体的复制或分裂的诱变剂。
3.针对不同作用方式的诱变剂,有不一样的致突变性检测方法。对于作用于dna的诱变剂来说,经常用到的检测试验方法有鼠伤寒沙门氏菌细菌回复突变试验(ames实验)以及哺乳动物细胞基因突变试验等。对于作用于染色体的诱变剂来说,经常用到的实验方法有染色体分析、染色体畸变实验、微核试验、显性致死实验、姐妹染色单体交换实验等。从2013年开始,欧盟规定,化妆品物质的致突变性基本测试,建议使用三种体外测试分析:ames测试、体外哺乳动物细胞突变试验、体外哺乳动物细胞微核试验或染色体畸变实验。
4.但使用实验的方法检测化学品致突变性有很多弊端,比如会花费大量的时间和精力等。近些年来计算机科学、化学信息学发展迅速,计算机建模成为辅助药物开发和预测毒性的有力工具。所以使用计算的方法预测化学品致突变性,逐渐发展起来。
5.目前已构建的致突变性预测模型虽然有其自身的特点,但也存在一些不足之处。这些不足主要体现在以下几个方面:第一,以往研究在使用计算的方法预测化学品致突变性时,建模使用的训练数据,多为单种实验数据或两种实验数据结合,没有覆盖检测所有致突变类型的实验,导致预测出的结果代表该种化学品是否仅作用于dna导致突变,或仅作用于染色体导致突变。预测结果存在片面性。第二,以往建立的预测模型大多未表征模型使用的应用域,导致在具体使用模型的过程中,对有些化学品致突变性的预测产生较大偏差。
6.基于以上原因,我搜集了包含ames实验、小鼠淋巴瘤试验、微核试验的实验数据,数据覆盖致使基因突变、染色体变异的实验结果。综合各实验结果,整理出涵盖三种实验结果的数据集。根据全面的数据集,基于python编程语言,使用机器学习方法建立预测化学品致突变性的模型,并表征应用域,明确模型的适用范围,使模型的使用范围更加明确。
技术实现要素:
7.本发明提供了一种简便、高效预测化学品致突变性的方法,该方法可以根据化合物的smiles码,预测其致突变性,为化学品风险评价和管理提供必要的基础依据。在建模过程中参照oecd对qsar模型构建和使用导则,进行了内、外部验证考察模型的预测能力和稳健性,并表征了模型的应用域,使模型更具备明确的应用范围。
8.本发明的技术方案如下:
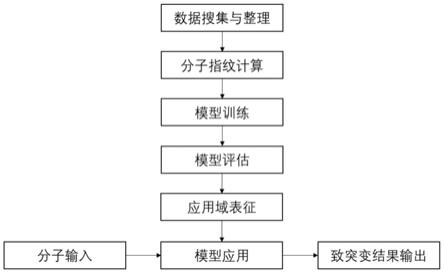
9.一种通过机器学习算法预测化学品致突变性的方法,步骤如下:
10.搜集并整理出6713种涵盖不同种实验的化合物致突变性数据,根据化合物的smiles码,计算其分子指纹;所有化合物的分子指纹及化合物的致突变性数据构成数据集;将数据集按照4:1比例随机拆分为训练集和验证集;训练集用于训练模型,验证集用于评估模型的外部预测能力;利用tanimoto系数结合阈值表征模型应用域;
11.(1)数据搜集与整理
12.(1.1)数据搜集:搜集不同化合物的体内微核实验数据、体外微核实验数据、ames实验数据和小鼠淋巴瘤实验数据及化合物对应的cas号和smiles码;若化合物有致突变性,实验数据为1,若化合物无致突变性,实验数据为0;
13.(1.2)数据综合:对于有多种实验的实验数据的化合物,若所有实验的实验数据一致,则保留该化合物及对应数据,若不同实验的实验数据不一致,则将该化合物及对应数据删除;
14.(1.3)数据处理:利用rdkit检查化合物对应smiles码的正确性,删除smiles码重复的化合物;同样利用rdkit对剩余化合物的smiles码进行中性化及脱盐处理,最终去除smiels码中含金属/非金属的化合物;
15.(2)计算化合物的分子指纹
16.(2.1)生成mol文件:利用rdkit中的molfromsmiles函数将化合物对应的smiles码转化为mol格式文件;
17.(2.2)计算分子指纹:利用rdkit中的getmorganfingerprintasbitvect函数,根据产生的mol文件计算morgan分子指纹;
18.(3)模型训练
19.将化合物的分子指纹及化合物的实验数据组合成数据集;将数据集按照4:1比例随机拆分为训练集和验证集,以训练集的实验数据1或0为因变量,以训练集的morgan分子指纹为自变量,选用梯度提升决策树算法,拟合因变量和自变量,训练模型;通过网格搜索法确定算法的最佳超参数,确定最佳超参数后,用模型拟合验证集的实验数据和morgan分子指纹;
20.最终确定出的模型超参数为:n_estimators=100,max_depth=26,min_samples_leaf=6,min_samples_split=100。
21.其中,n_estimators为最大的弱学习器的个数,max_depth为决策树最大深度,min_samples_leaf为叶子节点最少样本数,min_samples_split为内部节点再划分所需最小样本数。
22.(4)模型评估
23.通过5折交叉验证的方法计算模型在训练集的平均准确度,并通过roc曲线下方的面积大小:auc,来表征模型的效果;
24.使用验证集对模型进行外部验证,同样采用平均准确度和auc进行评价;
25.最终模型的评价结果为:
26.内部交叉验证准确率:0.789;内部交叉验证auc:0.863;外部验证准确率:0.799;外部验证auc:0.797。表明模型有较好的预测效果和稳定性。
27.(5)应用域表征
28.基于morgan分子指纹,使用rdkit中的tanimotosimilarity函数计算每个验证集分子与训练集分子之间的tanimoto系数t;
29.使用函数计算出的t评价两个分子之间的相似性:
[0030][0031]
其中,a和b是两种化学品的指纹;
[0032]
将阈值设定为0.25,若化合物与训练集中化合物分子相似性在0.25及以上的分子多于4个,则判定该化合物在应用域内,用此模型进行预测,否则认定该化合物在应用域外,不能用此模型进行预测。
[0033]
本发明的有利效果是:
[0034]
所建模型可以用于化合物的的多种类型的致突变性,且有明确的应用范围。该方法简便快捷、成本低廉。使用该发明专利的化合物致突变性预测结果,可以为化学品监管提供数据支持,对化学品的生态风险性评价具有重要意义。
附图说明
[0035]
图1为整体方法的构建流程。
具体实施方式
[0036]
以下结合附图和技术方案,进一步说明本发明的具体实施方式。
[0037]
实施例1
[0038]
给定一个化合物二亚硝基咖啡因(cas号:145438-97-7),要预测其致突变性,首先根据二亚硝基咖啡因的smiles码,利用rdkit软件包计算其分子指纹,然后计算其与训练集每个分子的相似性,计算得,训练集中分子与其相似性大于0.25的分子有5个,所以其在应用域内。根据其分子指纹,使用gbdt模型进行预测。得出结果为1,说明此化合物有致突变性。预测结果与实验结果相同。
[0039]
实施例2
[0040]
给定一个化合物对茴香胺(cas号:104-94-9),要预测其致突变性,首先根据对茴香胺的smiles码,利用rdkit软件包计算其分子指纹,然后计算其与训练集每个分子的相似性,计算得,训练集中分子与其相似性大于0.25的分子有267个,所以其在应用域内。根据其分子指纹,使用gbdt模型进行预测。得出结果为1,说明此化合物有致突变性。预测结果与实验结果相同。
[0041]
实施例3
[0042]
给定一个化合物10,10-二甲基十一烷-1-胺(cas号:68955-53-3),要预测其致突变性,首先根据10,10-二甲基十一烷-1-胺的smiles码,利用rdkit软件包计算其分子指纹,然后计算其与训练集每个分子的相似性,计算得,训练集中分子与其相似性大于0.25的分子有91个,所以其在应用域内。根据其分子指纹,使用gbdt模型进行预测。得出结果为0,说明此化合物无致突变性。预测结果与实验结果相同。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1