CD44介导的智能响应型聚合物胶束及其制备和应用
cd44介导的智能响应型聚合物胶束及其制备和应用
技术领域
1.本发明涉及胶束领域,尤其涉及一种cd44介导的智能响应型聚合物胶束及其制备和应用。
背景技术:
2.据统计,癌症已经成为人类死亡的主要原因之一。目前,药物治疗已经成为当今临床治疗肿瘤的重要手段。如今普遍的肿瘤治疗方法仍然以化疗手段为主,对患者的身体状况有不同程度的不良影响,容易出现呕吐、掉发等不良反应,易产生耐药性;且由于化疗药物缺乏对肿瘤的靶向性,使用过程中对正常细胞也有毒性。现已上市的白蛋白纳米粒等造价昂贵,肿瘤靶向性不理想,不能快速而有效的释放所负载的药物或基因。
3.胶束递送系统是一种最有潜力的纳米给药递送系统,在过去的二十年里作为抗肿瘤药物的递送载体得到了深度发展。与传统的抗肿瘤药物相比,载药聚合物胶束显示出多方面的优势,例如提高难溶性药物的溶解度,在到达靶向部位前增加药物在血液中的稳定性,通过增强渗透滞留(epr)效应显著提高药物在肿瘤组织的蓄积。因此,胶束递送系统由于能够持续增强治疗效果和减少系统毒性,而成为了一种有效的抗肿瘤药物递送系统。
4.迄今为止,胶束给药系统依然面临两个亟待解决的问题:
5.(1)肿瘤靶向效率和细胞内吞效率低下。一方面,epr效应仅实现纳米胶束在肿瘤部位的蓄积,而后续的入胞过程受胶束亲水外壳的影响,往往不尽如人意,这导致肿瘤细胞内药物浓度较低。
6.(2)药物的释放不受控制。入胞后,药物无法迅速从纳米胶束中释放并扩散至其靶部位,如细胞核。这些问题导致细胞内药物浓度过低而不能达到预期的抗肿瘤效果。
技术实现要素:
7.为解决上述技术问题,本发明的目的是提供一种cd44介导的智能响应型聚合物胶束及其制备和应用,本发明提供了基于聚合物组氨酸
‑
透明质酸
‑
烷基胺的cd44靶向胶束,用来递送抗肿瘤药物,由于胶束的ph敏感性,抗肿瘤药物可从聚合物胶束中释放。
8.本发明的第一个目的是提供一种ph响应性聚合物胶束,包括聚合物形成的胶束,聚合物包括透明质酸(ha),透明质酸通过化学键连接含10
‑
18个碳原子的烷基胺和组氨酸(his)。
9.进一步地,烷基胺选自十二胺(da)、癸胺、十四烷胺、十六烷胺和十八烷胺中的一种或几种。优选地,烷基胺为十二胺。烷基胺作为疏水端,其碳原子数目越多,疏水性越强,毒性越低,但其在透明质酸的接枝率越低,因此应选择合适碳原子数目的烷基胺。
10.进一步地,透明质酸上,烷基胺的接枝率为25%
‑
35%,组氨酸的接枝率为20%
‑
30%。
11.进一步地,透明质酸的分子量为8000
‑
12000。
12.优选地,聚合物为his
‑
ha
‑
da,包括ha,ha通过化学键连接da和his。
13.本发明的第二个目的是提供一种上述ph响应性聚合物胶束的制备方法,包括以下步骤:
14.(1)利用活化剂活化透明质酸,然后将活化的透明质酸与烷基胺在溶剂中于40
‑
55℃下反应,反应完全后得到接枝烷基胺的透明质酸;
15.(2)利用活化剂活化接枝烷基胺的透明质酸,然后与组氨酸在40
‑
55℃下反应,反应完全后得到聚合物;再将聚合物分散在水中,得到ph响应性聚合物胶束。
16.进一步地,在步骤(1)中,透明质酸和烷基胺的摩尔比为1:1。
17.进一步地,步骤(1)中的透明质酸和步骤(2)中的组氨酸的摩尔比为1:2。
18.进一步地,在步骤(1)和(2)中,活化剂为1
‑
(3
‑
二甲氨基丙基)
‑3‑
乙基碳二亚胺盐酸盐(edc.hcl)、n
‑
羟基丁二酰亚胺(nhs)和二环己基碳二亚胺中的一种或几种。
19.本发明以ha为骨架,在活化剂的催化下,连接烷基胺形成接枝烷基胺的透明质酸,其为两亲性聚合物;为得到具有ph敏感性的材料,通过具有咪唑基团的his对接枝烷基胺的透明质酸进行修饰,得到ph敏感型聚合物。
20.本发明的第三个目的是公开上述ph响应性聚合物胶束在制备抗肿瘤药物载体中的应用。
21.进一步地,抗肿瘤药物为疏水性抗肿瘤药物。
22.进一步地,抗肿瘤药物为阿霉素(dox)、蛇床子素、紫杉醇、去氢骆驼蓬、喜树碱、奥拉帕利、达沙替尼和依托泊苷中的一种或几种。
23.本发明的第四个目的是提供一种ph响应性聚合物载药胶束,其包括聚合物形成的胶束以及包载于胶束内的抗肿瘤药物,聚合物包括透明质酸(ha),透明质酸通过化学键连接含10
‑
18个碳原子的烷基胺和组氨酸(his)。
24.进一步地,ph响应性聚合物载药胶束的粒径为100
‑
150nm。
25.进一步地,ph响应性聚合物载药胶束的制备方法包括以下步骤:
26.将本发明的上述ph响应性聚合物胶束与抗肿瘤药物混合,得到ph响应性聚合物载药胶束。
27.本发明利用肿瘤细胞表面受体的目标配体进行修饰,使聚合物胶束具有主动靶向性,并通过配体
‑
受体介导的内吞作用显著增加细胞摄取量。在发明中,制备基于聚合物组氨酸
‑
透明质酸
‑
烷基胺的新型cd44靶向胶束,用来递送含显著毒副作用的化疗药物。ha为基本骨架,同时作为亲水段和活性靶向配体。为了制备亲水性聚合物,亲水段ha与疏水段烷基胺相连接形成透明质酸
‑
烷基胺,然后利用含有ph响应性基团的组氨酸修饰透明质酸
‑
烷基胺得到最后的聚合物组氨酸
‑
透明质酸
‑
烷基胺。在生理条件下,聚合物能够自组装形成胶束并包裹进疏水抗肿瘤药物。该聚合物胶束在血液循环中保持相对稳定,通过epr效应在肿瘤部位蓄积,从而通过cd44介导的内吞作用进入细胞。在细胞内含体/溶酶体中,由于胶束的ph敏感性,抗肿瘤药物从聚合物胶束中释放并逃出溶酶体。最后,游离的抗肿瘤药物扩散进入细胞核或其他靶部位达到抗肿瘤效果。
28.借由上述方案,本发明至少具有以下优点:
29.本发明基于生物制药技术和肿瘤微环境特征,提供了一种基于聚合物组氨酸
‑
透明质酸
‑
烷基胺且cd44介导的智能响应型聚合物胶束,该聚合物胶束可作为抗肿瘤药物载体,由于胶束的ph敏感性,抗肿瘤药物可从聚合物胶束中释放。其合成过程简便,有利于实
现肿瘤治疗的新突破。本发明以配体
‑
受体介导和抗增殖治疗为理论基础,针对受体过表达的肿瘤,发展主动靶向作用新型高效递药系统。
30.上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合详细附图说明如后。
附图说明
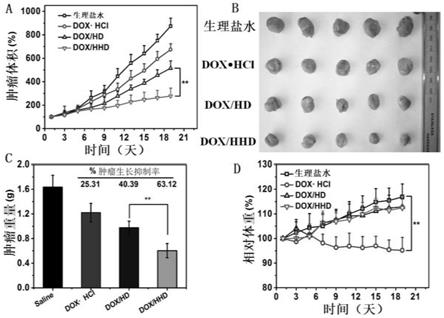
31.图1是ha、ha
‑
da和his
‑
ha
‑
da的1h
‑
nmr测试结果;
32.图2是his
‑
ha
‑
da在ph7.4和ph5.3下的临界胶束浓度(cmc)值;
33.图3是dox/hhd在ph5.3和7.4下的粒径分布图;
34.图4是dox/hhd在ph5.3和7.4下的zeta电位;
35.图5是dox/hhd在ph5.3和7.4下的包封率(ee%);
36.图6是dox
·
hcl、dox/hd和dox/hhd在ph5.3和7.4下的体外释放图;
37.图7是dox在不同剂型中的细胞摄取情况;
38.图8是不同胶束中dox的亚细胞分布情况测试结果;
39.图9是不同剂型dox下4t1细胞的存活率(a.24h,b.48h,n=5);
40.图10是胶束在小鼠体内的近红外荧光成像图;
41.图11是肿瘤组织的dir荧光强度;
42.图12是不同剂型dox在体内的分布情况;
43.图13是dox聚合物胶束体内抗肿瘤效果。
具体实施方式
44.下面结合实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
45.实施例1
46.1、ha
‑
da的合成
47.称取388mg(0.5mmol)的ha溶于30ml去离子水中,45℃下加入edc
·
hcl(192mg,1mmol)和nhs(115mg,1mmol)活化ha的羧基基团1h。称取92mg(0.5mmol)da溶于20ml无水乙醇中,缓慢滴加到ha溶液中,在45℃下反应24h。产物在蒸馏水中透析3
‑
4h除去副产物,冻干即得ha
‑
da。
48.2、his
‑
ha
‑
da的合成
49.称取194mg(0.5mmol)的ha
‑
da溶于10ml去离子水中,45℃下加入edc
·
hcl(96mg,0.5mmol)和nhs(57.5mg,0.5mmol)活化羧基30min,然后加入his(155mg,1mmol),在室温下反应12h。产物在蒸馏水中透析5
‑
6次,冻干即得his
‑
ha
‑
hd。
50.his
‑
ha
‑
hd的合成路线如下:
[0051][0052]
3、载药胶束的制备
[0053]
以ha
‑
hd和his
‑
ha
‑
hd为载体材料,分别利用透析法制备载药胶束dox/hd和dox/hhd。具体步骤如下:
[0054]
将5mg聚合物材料(ha
‑
hd或his
‑
ha
‑
hd)溶于5ml去离子水中得到1mg/ml的胶束溶液,依次向溶液中加入100μl三乙胺和0.5mldox
·
hcl溶液(5mg/ml),之后用透析袋在蒸馏水中透析4
‑
5次,透析后的液体用0.22μm孔径的微孔滤膜过滤,即得载药胶束。
[0055]
下面对该实施例中的化合物进行表征以及产物的性能测定:
[0056]
1、载体材料的结构表征
[0057]
取1
‑
2mg的聚合物(ha,ha
‑
da或his
‑
ha
‑
da)溶于适量d2o中通过1hnmr对其化学结构进行表征。如图1所示,δ3.0
–
4.0ppm代表ha骨架上h的化学位移,δ1.3ppm前后代表da上
‑
ch2上h原子的化学位移;δ7.2ppm代表his咪唑基团上h的化学位移。1hnmr结果表明两种聚合物已经成功合成,由特征峰的峰面积计算出his
‑
ha
‑
da中da和his的接枝率分别为32.5%和23.1%。
[0058]
2、his
‑
ha
‑
da在不同ph下的cmc值
[0059]
通过芘探针法测定his
‑
ha
‑
da空白胶束在不同ph下的cmc值。将芘溶液(10
‑6mol/l)加入离心管旋转蒸发除去有机溶剂,his
‑
ha
‑
da溶于pbs(ph 7.4或5.3)中得到浓度从0.39到100μg/ml的溶液,将该溶液加入上述离心管超声10min。密封、常温放置24h后,通过多功能酶标仪测量其荧光强度(λex=336nm,λem=373、384nm)。如图2所示,在生理条件下(ph 7.4),his
‑
ha
‑
da呈现出一个较低的cmc值6.37
×
10
‑4mg/ml,说明his
‑
ha
‑
hd在血液循环中能够抵制稀释自组装成聚合物胶束hhd,具有包载难溶性药物的潜能。然而,当ph下降到5.3时,hhd的cmc值增加到4.57
×
10
‑3mg/ml,表明his
‑
ha
‑
hd在酸性环境下难以形成聚合物胶束,其包载的药物可被迅速释放。
[0060]
3、载药胶束的质量评价
[0061]
载药胶束的粒径和zeta电位用动态激光粒径仪在不同ph下测得。包封率(ee%)在ph7.4下通过超滤法测得,取1ml的载药胶束置于超滤管中,3000r/min离心15min,通过多功能酶标仪测定下层溶液中的dox含量(λex=480nm,λem=590nm);载药量(dl%)测定是在载药胶束中加入过量乙醇,所包载的dox的量通过多功能酶标仪测得。式(1
‑
1)和式(1
‑
2)为计算ee%和dl%的公式。
[0062]
ee%=被包载药物浓度/总药物浓度
×
100%
ꢀꢀꢀ
(1
‑
1)
[0063]
dl%=被包载药物质量/(被包载药物质量+载体质量)
×
100%
ꢀꢀꢀ
(1
‑
2)
[0064]
如表1所示,dox/hd和dox/hhd有适宜的粒径、pdi和zeta电位。两种载药胶束均具有较高的ee%和dl%。dox/hd的粒径、zeta电位、ee%和dl%均与dox/hhd接近。相对来说,dox/hhd的粒径更小,粒径分布更加集中,这有助于实现epr效应。
[0065]
表1载dox胶束的表征
[0066][0067]
为了研究dox/hhd的ph响应性,测定其在不同ph下的粒径、zeta电位和dox的体外释放。如图3所示,与ph7.4下相比,dox/hhd在ph5.3下粒径明显增大。如图4所示,随着ph值从7.4降到5.3,dox/hhd表面的zeta电位约从
‑
25mv升到
‑
18mv。以上结果说明,在酸性环境下聚合物胶束的his段发生了质子化,导致胶束结构膨胀以及表面电荷增加。接着,测定在不同ph值下dox的荧光强度研究dox/hhd胶束的包封率。如图5所示,ph5.3下dox/hhd的包封率比ph 7.4下显著下降,说明在酸性环境下胶束结构不稳定,dox从胶束中泄露出来。
[0068]
4、载药胶束的体外释放
[0069]
通过透析法研究dox
·
hcl、dox/hd和dox/hhd的体外释放。取dox盐酸溶液(250μg/ml)、dox/hd和dox/hhd各2ml置于透析袋中(mwco=3500d),加入装有100ml pbs(ph5.3或7.4)的棕色瓶中,将棕色瓶置于37℃的恒温振荡箱中120r/min振荡,在预设时间点(0.5、1、2、4、6、8、12、24h)时取样1ml,样品通过多功能酶标仪(λex=480nm,λem=590nm)测定dox的浓度。如图6所示,ph7.4生理条件下,两种载药胶束均缓慢释药,而在ph5.3(溶酶体的弱酸性环境)下迅速释药;ph5.3下,dox/hhd的累计释放量达到了约70%,而dox/hd的累积释放量仅有约40%。结合前述实验结果分析,dox/hhd在生理条件下能够缓慢释药,减少dox的系统毒性,当到达肿瘤组织的弱酸性环境时,his上的咪唑基发生质子化,导致胶束结构不稳定而造成dox的释放。dox/hhd这种灵活的释药行为有利于减少药物到达靶向位点前的释放,增加肿瘤细胞的迅速释药。
[0070]
另外,对以上实施例制备的胶束进行以下细胞或活体实验:
[0071]
1.细胞摄取与亚细胞分布
[0072]
1.1细胞摄取
[0073]
dox胶束的细胞摄取量通过流式细胞仪测定。4t1细胞以105/孔的密度铺在六孔板中,孵育24h后,孔板中可另外加入游离的ha,或不加入游离的ha,加入不同剂型的dox(dox
为10μg/ml)孵育2h,之后吸出溶液用pbs(ph 7.4)清洗2
‑
3遍,通过流式细胞仪测定细胞内的dox浓度。如图7所示,其中图7(a)为使用共聚焦显微镜测定带有或不带有游离ha的dox负载胶束的细胞摄取情况。图7(b)为流式细胞术检测不同剂型dox的细胞摄取情况。
[0074]
1.2.亚细胞分布
[0075]
通过激光共聚焦显微镜考察dox在细胞内分布。将4t1细胞以105个/孔的密度铺到六孔板中,孵育24h后,盖玻片上细胞覆盖率约为80%。然后吸出培养基,用ph7.4的pbs清洗2
‑
3遍,加入1ml的pbs,将六孔板分为三组,分别加入dox、dox/hd、dox/hhd(dox为10μg/ml)孵育2h或者6h,吸出溶液,用pbs清洗2
‑
3遍,加入固定液(4%多聚甲醛)固定,10min后吸出、清洗,再加入染核试剂hoechst33258(1ml,10μg/ml),15min后吸出、清洗,取出盖玻片置于加有抗荧光猝灭剂的载玻片上,通过激光共聚焦显微镜观察。如图8所示,图8(a)为不同时间时dox/hhd或dox/hd在细胞内的分布情况;图8(b)为不同时间时空白hhd或hd胶束处理的细胞ao染色情况。
[0076]
2.mtt细胞毒性实验
[0077]
通过mtt法测定不同剂型dox的细胞毒性。将4t1细胞以3000个/孔的密度铺到96孔板中(200μl/孔),孵育24h后,吸出培养基,若需用ha预处理,每孔加入100μlha(1mg/ml,ph7.4)孵育2h,吸出溶液,按照预设的dox浓度梯度(20μg/ml到0.0390625μg/ml,最后一组为空白培养基)加入100μl的已过滤除菌的dox
·
hcl、dox/hd或dox/hhd,孵育48h后,每孔加入20μl的mtt溶液(5mg/ml,ph7.4)孵育4h,吸出孔内培养基,加入150μl的dmso,置于摇床上振荡10min,使结晶充分溶解,用酶联免疫检测仪在od490nm处测量各孔的吸光值。细胞存活率的计算公式如下:
[0078][0079]
其中,a490(treated)为加药孔的吸光度值,a490(non
‑
treated)为空白培养基孔的吸光度值,a0为无细胞孔的吸光度值。
[0080]
在ph7.4下,不同剂型dox对4t1细胞的毒性影响如图9所示,半数抑制浓度(ic50)如表2所示,三种剂型dox的细胞毒性具有浓度依赖性,与非敏感型胶束dox/hd相比,dox/hhd细胞毒性更高,ic50值更低,这可能是因为溶酶体中的低ph诱导了胶束结构变化,dox迅速释放,并且通过质子海绵效应逃出溶酶体。在用ha进行预处理后,载药胶束的细胞毒性受到抑制,表明dox/hhd是通过cd44受体介导的内吞作用进入细胞的,而预先加入的ha使细胞表面的cd44受体达到饱和,胞吞受抑。尽管dox
·
hcl的细胞摄取量较低,但细胞毒性却是最大的,这可能是dox
·
hcl与细胞核快速协同定位的原因。
[0081]
表2不同剂型dox对4t1细胞的半抑制浓度(ic50)
[0082][0083]
3.体内肿瘤靶向性实验
[0084]
3.1近红外荧光成像研究肿瘤靶向性
[0085]
肿瘤靶向性实验利用dir作为荧光探针通过近红外荧光成像系统进行研究。通过溶剂挥发法制得载dir胶束(dir/hd和dir/hhd),当肿瘤体积长到100mm3时,小鼠分别尾静脉注射0.1ml的dir/hd和dir/hhd(60μg/ml),在预设时间点(0、6、12、24、48h)通过近红外荧光成像系统考察dir在荷瘤小鼠体内的分布。48h后断颈处死小鼠,取出主要脏器(心肝脾肺肾)和肿瘤组织离体成像。如图10、11所示,dir/hd和dir/hhd在肿瘤部位均有明显的蓄积,表明粒径适宜,表面含ha的两种聚合物胶束通过epr效应以及ha
‑
cd44受体介导的主动靶向作用达到理想的体内肿瘤靶向性。然而,在预先注射0.1ml的ha(10mg/ml)后,dir胶束的肿瘤靶向性明显减弱,表明ha与肿瘤部位cd44受体的特异性结合是这些胶束产生靶向性的主要原因。两种聚合物胶束在肿瘤部位的荧光强度均随着时间的增加而增强,但dir/hhd组的荧光强度更高,表明hhd胶束有更好的肿瘤靶向性,可能是其ph敏感性导致的更好的肿瘤组织穿透性。另外,由于网状内皮系统(res)的捕获,在肝脏和脾脏中也有较强的dir荧光。
[0086]
3.2.dox的体内分布
[0087]
荷瘤小鼠尾静脉注射不同剂型的dox(dox为5mg/kg),24h后处死小鼠,取出肿瘤组织和主要脏器,加入1ml生理盐水匀浆,然后加入2ml有机溶剂(三氯甲烷:甲醇=3:1,v/v)进行萃取,离心(3000r/min,10min)取出有机层通过多功能酶标仪测量dox浓度。如图12所示,24h后dox在肿瘤部位有着明显的蓄积,其中,dox/hhd递送的dox量最多,与近红外荧光成像结果一致。dox
·
hcl组作为对照组,由于缺乏肿瘤靶向性,在肿瘤部位蓄积最少而在脏器中蓄积较多。
[0088]
4.体内药效学评价
[0089]
当荷瘤小鼠的肿瘤体积约为100mm3时,将小鼠分为四组,每组5只,在第1、5、9、13天时分别尾静脉注射0.1ml生理盐水、dox
·
hcl、dox/hd、dox/hhd(dox给药量为5mg/ml),给药期间,每2天测量小鼠的重量和肿瘤体积。第21天时,处死小鼠,取出肿瘤并称重。如图13所示,图13a为给药周期内肿瘤相对体积变化;图13b为21天时肿瘤取出图像;图13c为不同实验组的肿瘤重量;图13d为给药周期内小鼠体重变化。与生理盐水组相比,三种剂型的dox对肿瘤生长均有一定的抑制效果,其中,两种dox胶束的抗肿瘤效果比dox
·
hcl好。而在两种dox胶束中,由于肿瘤靶向性和细胞内ph敏感性释药的原因,ph敏感性dox/hhd胶束具有更高的肿瘤抑制率。此外,dox胶束产生的系统毒性比游离dox的小,注射载药胶束的两组小鼠几乎没有体重减轻。
[0090]
以上仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本技术领
域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1