基于病原微生物宏基因组的多种菌群间Granger因果分析方法与流程
本发明涉及高通量测序领域和分析技术领域,具体涉及基于病原微生物宏基因组的多种菌群间granger因果分析方法。
背景技术:
不明原因发热、疑难危重以及免疫缺陷等感染患者的诊断一直是困扰临床医生的难题,其病因常见三大类:感染、风湿免疫性疾病、恶性肿瘤等,这些患者普遍存在病原体诊断困难且多而复杂等问题,因病原体不能明确,治疗多限于经验性抗感染,不能精准施治。因此,准确、早期明确病原体并进行针对性抗感染治疗对这类患者预后至关重要。
基于高通量测序的宏基因组学(metagenomicnext-generationsequencing,mngs)可以检测样本中所有物种的dna或rna,能够快速分析患者样品中整体微生物群以及人类宿主基因组和转录组,因此,mngs对于发现新型病原体并检测健康和患病状态下的人类具有显著优势。文献“boulangecl,nevesal,chillouxjetal.impactofthegutmicrobiotaoninflammation,obesity,andmetabolicdisease[j].genomemed,2016,8(1):42.”运用高通量测序分析肠道微生物与肥胖、糖尿病和炎性肠病等疾病密切相关。文献“allegrettim,fabia,buglionis,martayanaetal.tearingdownthewalls:fdaapprovesnextgenerationsequencing(ngs)assaysforactionablecancergenomicaberrations[j].expclincancerres,2018,37(1):47.”研究发现mngs可鉴定与癌症相关的病毒,并用于晚期肿瘤的用药指导。因此以微生物为靶点,利用mngs干预微生物组必将成为临床治疗新的探索方向。
mngs技术的不断完善正在推动精准医学模式的发展,由于病原微生物宏基因组测序可检测范围包括基因组序列已知的6549种细菌,通常主要由宿主派生的序列和极小部分的病原体序列组成,为临床诊断构成了一个“大海捞针”的问题。因此背景微生物数据库(包括mngs测序过程中检测到的正常微生物和污染产生的微生物)的建立至关重要,我们对获取的病原微生物宏基因组数据先进行聚类分析划分出每一个菌种,进一步核算出每个菌种在疾病发展相应时间节点的物种丰度,对每个时间节点建立每组变量的时间序列并进行修正,最后对修正后的数据进行granger因果关系分析找到主要致病菌种,更快协助临床医师进行分析判断以实现临床诊断。
技术实现要素:
本发明利用高通量测序平台鉴定出多种细菌,然后结合granger因果分析方法找出这些细菌之间的因果作用关系,为从致病微生物样本中找出一种或几种致病菌提供一个快捷的鉴别方法,更快地协助临床医师进行分析判断以实现临床诊断。
为了达到上述技术效果,本发明通过以下技术方案实现的:
基于病原微生物宏基因组的多种菌群间granger因果分析方法,其原理为:对获取的病原微生物宏基因组数据进行聚类分析并划分出每一个菌种,核算出每个菌种在疾病发展相应时间节点的物种丰度,然后对每个时间节点建立每组变量的时间序列并进行修正,最后对修正后的数据进行granger因果分析,找到主要致病菌;
具体步骤如下:
s1:致病微生物样本的核酸提取;
s2:致病微生物核酸的二代高通量测序;
s3:测序原始数据的拼接与对比;
s4:根据otu聚类分析原理对每个菌种数据进行划分,otu聚类根据病原微生物基因数据库数据比对获得;
s5:统计每组数据在疾病发展过程中的时间节点以及对应的物种丰度,建立每个物种丰度的时间序列数据;
s6:使用matlab对每组数据的时间序列平稳性进行检验,筛选出若干组时间序列较为平稳的数据;
s7:利用二阶自回归模型对筛选出的数据作granger因果关系检验,得到每组数据相应的δ值;
s8:根据δ值的比较预测出主要致病菌。
本发明的有益效果是:利用高通量测序平台鉴定出多种细菌,然后结合granger因果分析方法找出这些细菌之间的因果作用关系,为从致病微生物样本中找出一种或几种致病菌提供一个快捷的鉴别方法,更快地协助临床医师进行分析判断以实现临床诊断;本发明提供的方法从统计学原理方面揭示病原微生物种群间的相互关系,以及挖掘主要致病微生物,通过理论分析结合最终实验结果出具的检验报告可以具有更高的可信度。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
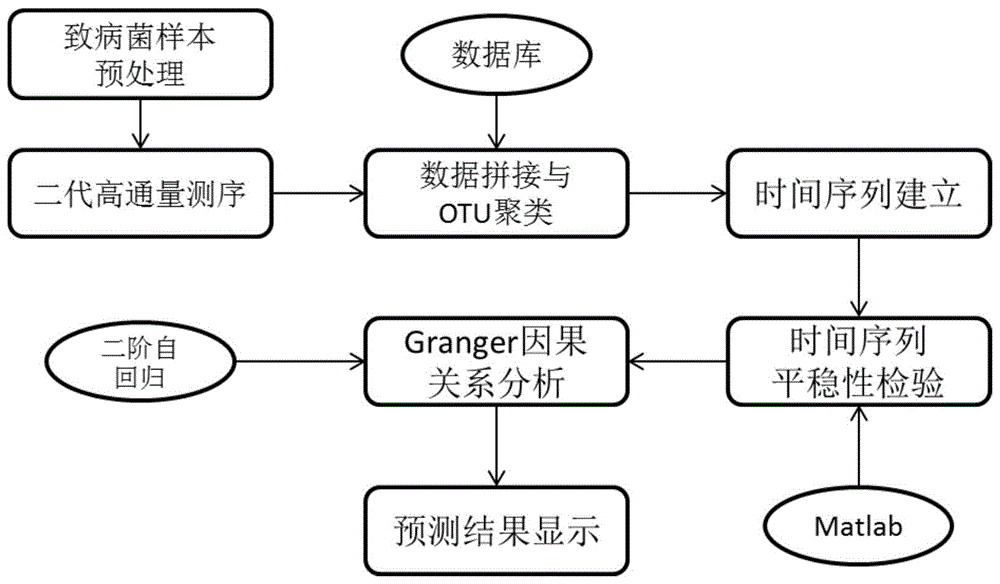
图1是本发明的多种菌群间granger因果分析方法的流程图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
如图1所示,本发明对获取的病原微生物宏基因组数据进行聚类分析并划分出每一个菌种,核算出每个菌种在疾病发展相应时间节点的物种丰度,然后对每个时间节点建立每组变量的时间序列并进行修正,最后对修正后的数据进行granger因果分析,找到主要致病菌;
具体步骤如下:
第一步:使用表面活性剂法将获得的致病微生物样本进行破碎;使用离心柱提取法对破碎的样本进行提取;使用磁珠分离法将提取出来的核酸进行纯化,得到高纯度的致病微生物核酸;
第二步:使用二代高通量测序仪将高纯度的致病微生物核酸进行测序;
第三步:使用flash对测序结果进行拼接和比对,获得目标基因序列;
第四步:使用seekdeep对目标基因序列进行out聚类分析,获得多个菌种;
第五步:统计每组数据在疾病发展过程中的时间节点以及对应的物种丰度,建立每个物种丰度的时间序列数据,针对微生物样本数据特点,设定时间序列x:{x1,x2,x3,......,xn},其中xn对应的是第n个样本的采样时间点,以时间轴t为横坐标求解自变量x,试图找到时间轴t同自变量x之间的关系,即x=f(t);
第六步:将每个物种丰度的时间序列数据导入matlab中,matlab对每组数据的时间序列平稳性进行检验,筛选出若干组时间序列较为平稳的数据;
第七步:利用二阶自回归模型对筛选出的数据作granger因果关系检验,得到每组数据相应的δ值;granger因果关系检验利用时间序列过去的a个点对当前的点求解回归方程,这里a为回归方程的阶,即lag,接下来通过时间范围t:lag上[x1lag]的点预测[lag+1]上的点[xlag+1],xlag+1-xplag+1产生误差ε1,以此类推时间范围t:(n-1)lag(n-1)上[xn-1log(n-1)]的点预测[n]上的点[xn],xn-xpn产生误差εn-lag。得到一系列的误差后,使用无偏估计方法求解联合回归产生的无偏估计误差δ;
第八步:通过比较两个时间序列的δ值而判定原因变量x和结果变量y,根据δ值的比较预测出主要致病菌。
在上述第六步中,基于基因数据采样时间点的不确定性,必须对样本的时间序列平稳性进行检验,并对达不到granger因果分析要求的数据在必要时进行修正。
在上述第七步中,基于宏基因数据的特点,对回归方程阶数的选择将影响granger因果分析的准确性。在数据量足够大的前提下,原则上要求阶数越大越好,但阶数越大,对计算性能要求就越高,因此需要合理选择。
术语解释
- 还没有人留言评论。精彩留言会获得点赞!