一种保元药物组合物的制备方法及其检测方法与流程
本发明涉及药物制备及检测领域,具体涉及一种保元药物组合物的制备方法及其检测方法。
背景技术:
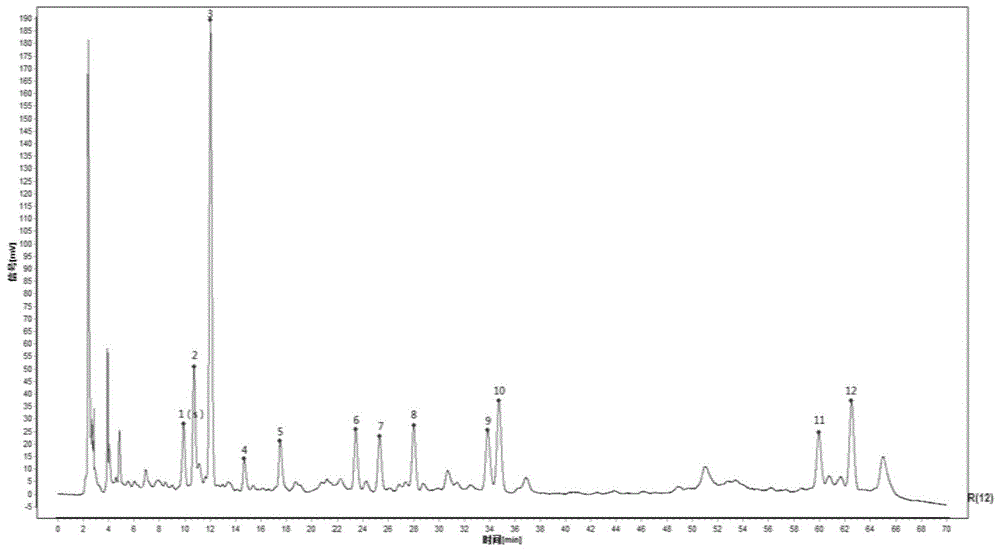
:保元汤出自《简明医彀》(明·孙志宏),“治元气虚弱,精神倦怠,肌肉柔慢,饮食少进,面青白,睡卧宁静,……及有杂证,皆属虚弱,宜服。”由人参、黄芪、甘草、肉桂四味药物组成。原文记载煎煮方法为:“人参一钱、黄芪二钱、甘草五分、肉桂二分,右加生姜一片,水煎服。”依据《历代度量衡》及《中国历代度、量、衡制演变简表》对保元汤中“制法及用法”中的数据单位同现代的数据单位进行换算。一钱为现今的3.69克,一分为0.37克,即保元汤煎煮工艺的“制法”可描述为取人参3.69g黄芪7.38g甘草1.85g肉桂0.74g,生姜1片,加水1920ml,浸泡30min,加热煎煮,滤弃药渣即得。根据经典名方目录中记载的方法,结合卫生部、国家中医药管理局《医疗机构中药煎药室管理规范》(国中医药发〔2009〕3号)进行煎煮制备“保元汤物质基准”,“保元汤物质基准”的制备应贴近传统、遵循古方原则,对本发明的制备方法及检测方法做进一步研究,以期进一步提高保元组合物有效成分的含量及科学的检测方法。基于上述原因,本发明以出膏率、人参皂苷rg1、rb1、re总量,黄芪甲苷含量、指纹图谱为指标,确定了药物组合物的前处理、加热方式、煎煮时间、滤过及干燥等最佳工艺相关参数,从而确定保元药物组合物的最佳制备方法,进一步提高了有效成分的含量以及产品的出膏率;通过对保元药物组合物检测方法进行摸索,确定了最佳的检测方法,更好的控制了产品的质量。技术实现要素:本发明的目的是提供一种保元药物组合物的制备方法。本发明的目的是提供一种保元药物组合物的检测方法。本发明所述的保元药物组合物的制备方法如下:人参60~85份,黄芪120~165份,甘草20~50份,肉桂10~20份,生姜15~25份,置于电子煎药罐中,加水1500~3020ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮0.5~2h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入药效上可接受的辅料制成药学上可接受的剂型。优选的,本发明所述的保元药物组合物的制备方法如下:人参74份,黄芪148份,甘草37份,肉桂15份,生姜20份,置于电子煎药罐中,加水1920ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.06mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-20~-45℃、冷冻干燥温度-50~-70℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入药效上可接受的辅料制成药学上可接受的剂型。本发明所述的药学上可接受的制剂为固体制剂或液体制剂。本发明所述固体制剂为颗粒剂、胶囊剂、片剂、丸剂、散剂、冻干粉针剂。本发明所述液体制剂为注射制剂、口服液。本发明所述辅料没有限制,药学上能接受即可。本发明所述生姜一片以3g计。本发明所述检测方法为:性状:本品为棕色至棕褐色粉末,气微香,味微苦甜;鉴别:1)取本品粉末0.5~1.5g,加三氯甲烷20~60ml,加热回流0.5~2小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理20~40分钟,吸取上清液加2~4倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取人参对照药材1g,同法制成对照药材溶液;再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rf对照品及人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液、对照药材溶液、对照品溶液各1~2μ1,分别点于同一硅胶g薄层板上,以比例为15:40:22:10的三氯甲烷-乙酸乙酯-甲醇-水6~15℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和365nm紫外光灯下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的斑点,阴性色谱在对照色谱相应的位置上无斑点;2)取本品1~2g,加水15~35ml,超声处理20~40分钟,滤过,滤液用水饱和正丁醇振摇提取2~4次,每次20~30ml,合并正丁醇提取液,用氨试液洗涤2~4次,每次20~40ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液;另取黄芪对照药材1~2g,加水40~60ml,煎煮20~40分钟,滤过,滤液同法制成对照药材溶液;再取黄芪甲苷对照品,加甲醇制成每lml含2mg的溶液,作为对照品溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述对照品溶液、对照药材溶液、供试品溶液各10μl,分别点于同一硅胶g薄层板上,以比例为13:7:2三氯甲烷-甲醇-水5~15℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;置365nm紫外光灯下显检视,显相同颜色的荧光斑点;3)取本品及缺甘草阴性样品各0.5~1.5g,加水20~60ml,超声20~40分钟,滤过,用乙醚振摇提取2~4次,每次10~30ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取2~4次,每次15~25ml,合并正丁醇液,用氨试液振摇提取2~4次,每次20~40ml,合并氨试液,用盐酸调ph值至3~4;再用乙酸乙酯振摇提取2~4次,每次20~40ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液及缺甘草阴性样品溶液;另取炙甘草饮片0.5~1.5g,加水40~60ml,煎煮20~40分钟,滤过,同法制成对照药材溶液;取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述供试品溶液及缺甘草阴性样品溶液、对照药材溶液、对照品溶液各5~10μl,分别点于同一硅胶g薄层板上,以比例为13:7:2的三氯甲烷-甲醇-水5~10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及365nm紫外光灯下检视,供试品色谱中,在与对照品、对照药材色谱相应的位置上,显相同颜色的斑点或荧光斑点;4)取本品及缺肉桂阴性冻干粉各0.5~1.5g,加水5~15ml使其溶解,用乙醚振摇提取1~3次,每次15~35ml,合并乙醚液,挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液;另取肉桂饮片0.5~1.5g,加水40~60ml,煎煮20~40分钟,滤过,同法制成对照药材溶液;再取肉桂酸对照品,用40~65%甲醇制成1ml含0.2mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取吸取上述供试品溶液及缺肉桂阴性样品溶液、对照药材溶液、对照品溶液各3μl,点于同一硅胶gf254薄层板上,以比例为5:5:0.3环己烷-乙醚-冰醋酸为展开剂,展开,取出,晾干,置254nm紫外光灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点或荧光斑点;检查:水分照(《中国药典》2015版四部通则0832)第二法测定不得过12.0%;浸出物:照醇溶性浸出物测定法(《中国药典》2015版四部通则2201)项下热浸法测定,浸出物为18%~81.0%;指纹图谱:照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;色谱柱:inertsustain-aq-c18,以乙腈为流动相a,以0.1%磷酸溶液为流动相b,流速为1ml/min,检测波长为200~220nm,具体流动相梯度洗脱程序为:流动相梯度洗脱程序参照物溶液的制备取毛蕊异黄酮葡萄糖苷对照品适量,精密称定,加40~60%甲醇制成每1ml含毛蕊异黄酮葡萄糖苷50.4592μg/ml的溶液,即得;供试品溶液的制备取本品1.5~3.5g,精密称定,置50ml三角锥瓶中,加40~60%甲醇40~60ml超声30~60min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取2~3次,每次15~35ml,合并正丁醇溶液,用20~40ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加40~60%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得;测定法分别精密吸取参照物溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录70min的色谱图,即得;供试品指纹图谱中应分别呈现相应的参照物色谱峰保留时间相同的色谱峰;按中药色谱指纹图谱相似度评价系统计算,供试品指纹图谱与对照指纹图谱的相似度不得低于0.85,对照指纹图谱见图1;含量测定:人参照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为190~210nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000;按规定梯度洗脱程序进行梯度洗脱,流动相梯度洗脱程序为:流动相梯度洗脱程序对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得;供试品溶液的制备取本品0.5~1.5g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取2~4次,每次10~20ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取2~4次,每次20~40ml,合并正丁醇液,用氨试液洗涤2~4次,每次20~40ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液10~30ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得;测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得;本品按干燥品计算,每1g含人参以人参皂苷rg1、rb1、re的总含量计,应为0.6mg/g~2.4mg/g;含量测定黄芪照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验十八烷基硅烷键合硅胶为填充剂;以乙腈-水(35:65)为流动相;蒸发光散射检测器检测;理论板数按黄芪甲苷峰计算应不低于4000;对照品溶液的制备取黄芪甲苷对照品适量,精密称定,加甲醇制成每lml含0.5mg的溶液,即得;供试品溶液的制备取本品0.5~1.5g,精密称定,精密加水40~60ml,称定重量,超声处理(功率720w,频率40khz)30~60分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取2~6次,每次20~40ml,合并正丁醇提取液,用氨试液洗涤2~4次,每次20~40ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。本品按干燥品计算,每1g含黄芪以黄芪甲苷(c41h68o14)计,应为0.10mg/g~1.40mg/g。优选的,本发明所述检测方法为:1)取本品粉末1g,加三氯甲烷40ml,加热回流1小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理30分钟,吸取上清液加3倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取人参对照药材1g,同法制成对照药材溶液;再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rf对照品及人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液;照薄层色谱法(通则0502)试验,吸取供试品溶液、对照药材溶液、对照品溶液各1~2μ1,分别点于同一硅胶g薄层板上,以比例为15:40:22:10的三氯甲烷-乙酸乙酯-甲醇-水10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和365nm紫外光灯下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的斑点,阴性色谱在对照色谱相应的位置上无斑点;2)取本品1.5g,加水25ml,超声处理30分钟,滤过,滤液用水饱和正丁醇振摇提取2次,每次25ml,合并正丁醇提取液,用氨试液洗涤2次,每次30ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液;另取黄芪对照药材1.5g,加水50ml,煎煮30分钟,滤过,滤液同法制成对照药材溶液;再取黄芪甲苷对照品,加甲醇制成每lml含2mg的溶液,作为对照品溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取对照品溶液、对照药材溶液、供试品溶液各10μl,分别点于同一硅胶g薄层板上,以比例为13:7:2三氯甲烷-甲醇-水10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;置365nm紫外光灯下显检视,显相同颜色的荧光斑点;3)取本品及缺甘草阴性样品各1g,加水40ml,超声30分钟,滤过,用乙醚振摇提取2次,每次20ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取3次,每次20ml,合并正丁醇液,用氨试液振摇提取2次,每次30ml,合并氨试液,用盐酸调ph值至3~4;再用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液及缺甘草阴性样品溶液;另取炙甘草饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液;取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液;照薄层色谱法(通则0502)试验,吸取供试品溶液及缺甘草阴性样品溶液、对照药材溶液、对照品溶液各10μl,分别点于同一硅胶g薄层板上,以比例为13:7:2的三氯甲烷-甲醇-水5~10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及365nm紫外光灯下检视,供试品色谱中,在与对照品、对照药材色谱相应的位置上,显相同颜色的斑点或荧光斑点;4)取本品及缺肉桂阴性冻干粉各1g,加水10ml使其溶解,用乙醚振摇提取1次,每次25ml,将乙醚液挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液;另取肉桂饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液;再取肉桂酸对照品,用50%甲醇制成1ml含0.2mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取供试品溶液及缺肉桂阴性样品溶液、对照药材溶液、对照品溶液各3μl,点于同一硅胶gf254薄层板上,以比例为5:5:0.3环己烷-乙醚-冰醋酸为展开剂,展开,取出,晾干,置254nm紫外光灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点或荧光斑点;优选的,本发明所述指纹图谱检测方法为:指纹图谱:照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;色谱柱:inertsustain-aq-c18,以乙腈为流动相a,以0.1%磷酸溶液为流动相b,流速为1ml/min,检测波长为210nm,具体流动相梯度洗脱程序为:流动相梯度洗脱程序参照物溶液的制备取毛蕊异黄酮葡萄糖苷对照品适量,精密称定,加50%甲醇制成每1ml含毛蕊异黄酮葡萄糖苷50.4592μg/ml的溶液,即得;供试品溶液的制备取本品2.5g,精密称定,置50ml三角锥瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得;测定法分别精密吸取参照物溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录70min的色谱图,即得;供试品指纹图谱中应分别呈现相应的参照物色谱峰保留时间相同的色谱峰;按中药色谱指纹图谱相似度评价系统计算,供试品指纹图谱与对照指纹图谱的相似度不得低于0.85,对照指纹图谱见图1;优选的,本发明所述人参含量测定方法为:人参照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为203nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000;按规定梯度洗脱程序进行梯度洗脱,梯度洗脱程序为:梯度洗脱程序对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得;供试品溶液的制备取本品1g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得;测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得;本品按干燥品计算,每1g含人参以人参皂苷rg1、rb1、re的总含量计,应为0.6mg/g~2.4mg/g;优选的,本发明所述黄芪含量测定方法为:含量测定黄芪照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验十八烷基硅烷键合硅胶为填充剂;以乙腈-水(35:65)为流动相;蒸发光散射检测器检测;理论板数按黄芪甲苷峰计算应不低于4000;对照品溶液的制备取黄芪甲苷对照品适量,精密称定,加甲醇制成每lml含0.5mg的溶液,即得;供试品溶液的制备取本品1.0g,精密称定,精密加水50ml,称定重量,超声处理45分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取4次,每次25ml,合并正丁醇提取液,用氨试液洗涤3次,每次30ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。本品按干燥品计算,每1g含黄芪以黄芪甲苷(c41h68o14)计,应为0.10mg/g~1.40mg/g。有益效果:本发明与现有技术相比,具有以下有益效果:1、本发明以出膏率、人参皂苷rg1、rb1、re总量,黄芪甲苷含量、指纹图谱为考察指标,确定了药物组合物的前处理、加热方式、煎煮时间、滤过及干燥等最佳工艺相关参数,从而确定保元药物组合物的最佳制备方法,进一步提高了有效成分的含量以及产品的出膏率。通过对保元药物组合物检测方法进行摸索,确定了最佳的检测方法,更好的控制了产品的质量。2、本发明从人参皂苷rg1、rb1、re的总量、黄芪甲苷总量上分析,三批验证提取液与干膏粉数据均基本平行,且工艺过程中物质传递损失较小;从浸出物、水分上分析,三批验证干膏粉无明显差异。从指纹图谱上分析,峰2、3、6、10为甘草色谱峰,峰1、4、5、8、11、12为黄芪色谱峰。暂以1号峰为参照峰计算相对保留时间,结果表明、药材、饮片、提取液、干膏粉色谱图中共有峰,其相对保留时间rsd小于2.0%,故所确定的物质基准制备工艺稳定可行。3、本发明通过工艺研究考察明火及电子煎药罐两种加热方式,结果从煎煮时间、出膏率、人参皂苷rg1、rb1、re总量,黄芪甲苷含量综合分析,两种加热方式差异不大,由于电子煎药罐更为便捷,故选择加热方式为电子煎药罐。对煎煮时间代替提取液体积作为煎煮终点进行研究,确定保元组合物物质基准煎煮时间为30-60min,优选为60min,提取液人参皂苷rg1、rb1、re总含量0.106mg/ml;提取液黄芪甲苷含量0.031mg/g;转移率人参皂苷rg1、rb1、re总含量27.32%;转移率黄芪甲苷总含量最高达到了24.86%;含固率0.064g/ml。4、本发明混合药材的浸泡时间在40min后吸水重量趋于稳定,故将保元汤药材的最佳提取浸泡时间定为40min。5、本发明由加水量因素考察可知,确定保元药物组合物物质基准的加水量为1920-3020ml,最佳煎煮加水量为7倍量水,即1920ml。提取液人参皂苷rg1、rb1、re总含量0.078mg/g;提取液含量13.61mg/g;转移率人参皂苷rg1、rb1、re总含量17.70%;转移率黄芪甲苷总含量最高达到了26.25%;含固率0.065g/ml。6、本发明通过过滤实验,过滤方法优先选用200目滤布过滤,澄明度澄清。7、本发明通过浓缩方法考察,对常压浓缩与真空浓缩相比较,优先采用真空的方法进行浓缩,人参皂苷rg1、rb1、re总含量最高达0.351mg/ml;提取液黄芪甲苷含量最高可达0.089mg/g;转移率人参皂苷rg1、rb1、re总含量17.07%;转移率黄芪甲苷含量最高达到了23.10%;含固率0.235g/ml。冻干粉不发泡不融化、干膏粉质地疏松。8、本发明通过浓缩真空度考察,确定了浓缩真空度为:-0.08mpa~-0.04mpa,最佳真空度为-0.08mpa~-0.06mpa的方法进行浓缩。人参皂苷rg1、rb1、re总含量最高达0.390mg/ml;提取液黄芪甲苷含量最高可达0.103mg/g;转移率人参皂苷rg1、rb1、re总含量18.98%;转移率黄芪甲苷含量最高达到了27.14%;含固率0.248%。9、本发明通过浓缩温度考察,确定浓缩温度为46-60℃,最佳浓缩温度为55℃~60℃,人参皂苷rg1、rb1、re总含量最高达0.487mg/ml;提取液黄芪甲苷含量最高可达0.098mg/g;转移率人参皂苷rg1、rb1、re总含量20.35%;转移率黄芪甲苷含量最高达到了25.06%;含固率0.244%。10、本发明通过工艺验证测定,人参皂苷rg1、rb1、re的总量含量2.306mg/g,黄芪甲苷含量0.890mg/g。11、本发明通过对人参【鉴别】项摸索试验,人参皂苷rg1、rb1、re对照品溶液与人参饮片点样量优选均为10μl,供试品溶液点样量为10μl,相应位置下的显色斑点均清晰可见,没有出现拖尾现象。耐用性考察,通过对低温低湿、常温、高温高湿环境及不同厂家的硅胶g薄层板进行耐用性考察,结果不同影响因素下,此人参薄层鉴别条件耐用性良好。12、本发明通过对黄芪【鉴别】项摸索试验,黄芪甲苷对照品溶液与黄芪饮片点样量优选均为10μl,供试品溶液点样量分别为5μl,8μl和10μl,相应位置下的显色斑点均清晰可见,没有出现拖尾现象,耐用性考察,通过对低温低湿、常温、高温高湿环境及不同厂家的硅胶g薄层板进行耐用性考察,结果不同影响因素下,此黄芪薄层鉴别条件耐用性良好。13、本发明通过对甘草【鉴别】项摸索试验,供试品色谱中,在与甘草对照药材和对照品色谱相应的位置上,显相同颜色的斑点,无斑点拖尾严重,故确定供试品溶液和对照品、对照药材溶液的点样量优选均为10μl。通过对低温低湿、常温、高温环境进行耐用性考察,结果不同影响因素下,此薄层鉴别条件耐用性良好。14、本发明通过对肉桂【鉴别】项摸索试验,供试品色谱中,在与肉桂对照药材和对照品色谱相应的位置上,显相同颜色的斑点,无斑点拖尾严重,故确定供试品溶液和对照品、对照药材溶液的点样量优选均为10μl。通过对低温低湿、常温、高温环境进行耐用性考察,结果不同影响因素下,此薄层鉴别条件耐用性良好。15、本发明通过指纹图研究,确定了指纹图谱的检测方法,图谱分离度好,杂质峰少。通过方法学考察,空白溶剂图谱和供试品溶液色谱峰的比较发现空白无干扰,仪器精密度、稳定性、重复性良好。附图说明图1对照指纹图谱(峰1(s):毛蕊异黄酮葡萄糖苷峰11:甘草苷酸铵)。图2保元汤物质基准工艺验证指纹图谱对比图(1、byt-2019102401t;2、byt-2019102402t;3、byt-2019102403t;4、byt-2019102401n;5、byt-2019102402n;6、byt-2019102403n;7、byt-2019102401d;8、byt-2019102402d;9、byt-2019102403d;10、甘草药材溶液;11、甘草酸铵对照品;12、黄芪饮片溶液;13、毛蕊异黄酮葡萄糖苷)图3药材-饮片-提取液-物质基准量值传递叠加图(从下至上:物质基准d-181016-01、浓缩液n-181016-01、提取液t-181016-01、甘草饮片gc-zy-2017110301y、黄芪饮片bs-mc-2017112201y)图4不同批次对应实物(冻干粉)性状图5指纹图谱色谱图-1图6指纹图谱色谱图-2图7指纹图谱色谱图-3图8指纹图谱色谱图-4图9指纹图谱色谱图-5图10不同提取溶媒指纹图谱测定结果(s1:50%甲醇提取;s2:水饱和的正丁醇溶液萃取;s3:乙酸乙酯萃取)图11指纹图谱饮片归属色谱图(s1:人参;s2:黄芪饮片;s3:甘草;s4:肉桂;s5:毛蕊异黄酮葡萄糖苷;s6:供试品;s7:甘草酸铵;s8:空白溶剂)图12整体性考察色谱图(s1:供试品溶液;s2:毛蕊异黄酮葡萄糖苷对照品溶液;s3:空白溶剂)图13指纹图谱精密度测定色谱图图14指纹图谱稳定性测定色谱图(s1:byt-2019091604d-0h;s2:byt-2019091604d-3h;s3:byt-2019091604d-6h;s4:byt-2019091604d-9h;s5:byt-2019091604d-12h;s6:byt-2019091604d-24h)图15指纹图谱重复性测定色谱图(s1:byt-2019091604d-1;s2:byt-2019091604d-2;s3:byt-2019091604d-3;s4:byt-2019091604d-4;s5:byt-2019091604d-5;s6:byt-2019091604d-6)图16不同批次物质基准指纹图谱共有模式图-1(r、对照图谱;s1、byt-2019091001d;s2、byt-2019091002ds;3、byt-2019091003d;s4、byt-2019091004d;s5、byt-2019091005d;s6、byt-2019091606d;s7、byt-2019091807d;s8、byt-2019091008d;s9、byt-2019091009d;s10、byt-2019092310d;s11、byt-2019092311d;s12、byt-2019092312d;s13、byt-2019092413d;s14、byt-2019092414d;s15、byt-2019092415d)具体实施方式下面通过具体实施例,对本发明的技术方案作进一步地具体说明。实施例1制备方法人参74g,黄芪148g,甘草37g,肉桂15g,生姜20片,置于电子煎药罐中,加水1920ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.06mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-20~-45℃、冷冻干燥温度-50~-70℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,制成颗粒,即得颗粒剂。实施例2制备方法人参74g,黄芪148g,甘草37g,肉桂15g,生姜20片,置于电子煎药罐中,加水1920ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.06mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-20~-45℃、冷冻干燥温度-50~-70℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,装入胶囊,即得胶囊剂。实施例3制备方法人参74g,黄芪148g,甘草37g,肉桂15g,生姜20片,置于电子煎药罐中,加水1920ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.06mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-20~-45℃、冷冻干燥温度-50~-70℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,压片,即得片剂。实施例4制备方法人参74g,黄芪148g,甘草37g,肉桂15g,生姜20片,置于电子煎药罐中,加水1920ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.06mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-20~-45℃、冷冻干燥温度-50~-70℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,干燥,制成丸剂。实施例5制备方法人参74g,黄芪148g,甘草37g,肉桂15g,生姜20片,置于电子煎药罐中,加水1920ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.06mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-20~-45℃、冷冻干燥温度-50~-70℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入15倍量的纯静用水,混匀,过滤,灭菌,装瓶,得口服液。实施例6制备方法人参74g,黄芪148g,甘草37g,肉桂15g,生姜20片,置于电子煎药罐中,加水1920ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.06mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-20~-45℃、冷冻干燥温度-50~-70℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入28倍量的注射用水,混匀,过滤,灭菌,得注射剂。实施例7制备方法人参60g,黄芪120g,甘草20g,肉桂10g,生姜20片,置于电子煎药罐中,加水1500ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮0.5h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,制成颗粒,即得颗粒剂。实施例8制备方法人参60g,黄芪120g,甘草20g,肉桂10g,生姜20片,置于电子煎药罐中,加水1500ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮0.5h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,即得胶囊剂。实施例9制备方法人参60g,黄芪120g,甘草20g,肉桂10g,生姜20片,置于电子煎药罐中,加水1500ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮0.5h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,即得片剂。实施例10制备方法人参60g,黄芪120g,甘草20g,肉桂10g,生姜20片,置于电子煎药罐中,加水1500ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮0.5h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,干燥,制成丸剂。实施例11制备方法人参60g,黄芪120g,甘草20g,肉桂10g,生姜20片,置于电子煎药罐中,加水1500~3020ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮0.5~2h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入15倍量的纯静用水,混匀,过滤,灭菌,装瓶,得口服液。实施例12制备方法人参60g,黄芪120g,甘草20g,肉桂10g,生姜20片,置于电子煎药罐中,加水1500ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮0.5h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入28倍量的注射用水,混匀,过滤,灭菌,得注射剂。实施例13制备方法人参85g,黄芪165g,甘草50g,肉桂20g,生姜20片,置于电子煎药罐中,加水3020ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮2h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,制成颗粒,即得颗粒剂。实施例14制备方法人参85g,黄芪165g,甘草50g,肉桂20g,生姜20片,置于电子煎药罐中,加水3020ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮2h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,装入胶囊,即得胶囊剂。实施例15制备方法人参85g,黄芪165g,甘草50g,肉桂20g,生姜20片,置于电子煎药罐中,加水3020ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮2h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,压片,即得片剂。实施例16制备方法人参85g,黄芪165g,甘草50g,肉桂20g,生姜20片,置于电子煎药罐中,加水3020ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮2h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入可溶性淀粉(可溶性淀粉与干膏比是1:1~1:4)混合均匀,干燥,制成丸剂。实施例17制备方法人参85g,黄芪165g,甘草50g,肉桂20g,生姜20片,置于电子煎药罐中,加水3020ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮2h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入15倍量的纯静用水,混匀,过滤,灭菌,装瓶,得口服液。实施例18制备方法人参85g,黄芪165g,甘草50g,肉桂20g,生姜20片,置于电子煎药罐中,加水3020ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮0.5~2h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.04mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-15~-50℃、冷冻干燥温度-45~-75℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得,或收集干膏粉后加入28倍量的注射用水,混匀,过滤,灭菌,得注射剂。实施例1-18按以下任一检测方法检测实施例19检测方法【性状】本品为棕色至棕褐色粉末,气微香,味微苦甜。【鉴别】(1)取本品粉末1g,加三氯甲烷40ml,加热回流1小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理30分钟,吸取上清液加3倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取人参对照药材1g,同法制成对照药材溶液。再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rf对照品及人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述供试品溶液、对照药材溶液、对照品溶液各1~2μ1,分别点于同一硅胶g薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和紫外光灯(365nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的斑点,阴性色谱在对照色谱相应的位置上无斑点。(2)取本品1.5g,加水25ml,超声处理30分钟,滤过,滤液用水饱和正丁醇振摇提取2次,每次25ml,合并正丁醇提取液,用氨试液洗涤2次,每次30ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液。另取黄芪对照药材1.5g,加水50ml,煎煮30分钟,滤过,滤液同法制成对照药材溶液。再取黄芪甲苷对照品,加甲醇制成每lml含2mg的溶液,作为对照品溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取对照品溶液、对照药材溶液、供试品溶液各10μl,分别点于同一硅胶g薄层板上,以三氯甲烷_甲醇-水(13:7:2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;置紫外光灯(365nm)下显检视,显相同颜色的荧光斑点。(3)取本品及缺甘草阴性样品各1g,加水40ml,超声30分钟,滤过,用乙醚振摇提取2次,每次20ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取3次,每次20ml,合并正丁醇液,用氨试液振摇提取2次,每次30ml,合并氨试液,用盐酸调ph值至3~4。再用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取(炙)甘草饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述供试品溶液、缺甘草阴性样品溶液、对照药材溶液、对照品溶液各10μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水(13:7:2)5~10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及紫外光灯(365nm)下检视,供试品色谱中,在与对照品、对照药材色谱相应的位置上,显相同颜色的斑点或荧光斑点。(4)取本品及缺肉桂阴性冻干粉各1g,加水10ml使其溶解,用乙醚振摇提取1次,每次25ml,合并乙醚液,挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液。另取肉桂饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。再取肉桂酸对照品,用50%甲醇制成1ml含0.2mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述吸取上述供试品溶液、缺肉桂阴性样品溶液、对照药材溶液、对照品溶液各3μl,点于同一硅胶gf254薄层板上,以环己烷-乙醚-冰醋酸(5:5:0.3)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点或荧光斑点;【检查】水分不得过12.0%(通则0832第二法)。【浸出物】照醇溶性浸出物测定法(《中国药典》2015版四部通则2201)项下热浸法测定,浸出物为18%~81.0%。【指纹图谱】照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;色谱柱:inertsustain-aq-c18,以乙腈为流动相a,以0.1%磷酸溶液为流动相b,流速为1ml/min,检测波长为210nm,流动相梯度洗脱程序如下。流动相梯度洗脱程序参照物溶液的制备取毛蕊异黄酮葡萄糖苷对照品适量,精密称定,加50%甲醇制成每1ml含毛蕊异黄酮葡萄糖苷50.4592μg/ml的溶液,即得。供试品溶液的制备取本品约2.5g,精密称定,置50ml三角锥瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得。测定法分别精密吸取参照物溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录70min的色谱图,即得。供试品指纹图谱中应分别呈现相应的参照物色谱峰保留时间相同的色谱峰。按中药色谱指纹图谱相似度评价系统计算,供试品指纹图谱与对照指纹图谱的相似度不得低于0.85,对照指纹图谱见图1。【含量测定】人参照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为203nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000。按以下规定的流动相梯度洗脱程序洗脱。流动相梯度洗脱程序对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得供试品溶液的制备取本品约1g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得。本品按干燥品计算,每1g含人参以人参皂苷rg1(c42h72o14)、rb1(c54h92o23)、re(c48h82o18)的总含量计,应为0.6mg/g~2.4mg/g。【含量测定】黄芪照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。色谱条件与系统适用性试验十八烷基硅烷键合硅胶为填充剂;以乙腈-水(35:65)为流动相;蒸发光散射检测器检测。理论板数按黄芪甲苷峰计算应不低于4000。对照品溶液的制备取黄芪甲苷对照品适量,精密称定,加甲醇制成每lml含0.5mg的溶液,即得。供试品溶液的制备取本品1.0g,精密称定,精密加水50ml,称定重量,超声处理(功率720w,频率40khz)45分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取4次,每次25ml,合并正丁醇提取液,用氨试液洗涤3次,每次30ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。本品按干燥品计算,每1g含黄芪以黄芪甲苷(c41h68o14)计,应为0.10mg/g~1.40mg/g。【包装】真空包装袋密封。【贮存】置阴凉干燥处贮存。实施例20检测方法【性状】本品为棕色至棕褐色粉末,气微香,味微苦甜;【鉴别】(1)取本品粉末0.5g,加三氯甲烷20ml,加热回流0.5小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理20分钟,吸取上清液加2倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取人参对照药材1g,同法制成对照药材溶液;再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rf对照品及人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液、对照药材溶液、对照品溶液各1~2μ1,分别点于同一硅胶g薄层板上,以比例为15:40:22:10的三氯甲烷-乙酸乙酯-甲醇-水6~15℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和365nm紫外光灯下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的斑点,阴性色谱在对照色谱相应的位置上无斑点;(2)取本品1g,加水15ml,超声处理20分钟,滤过,滤液用水饱和正丁醇振摇提取2次,每次20ml,合并正丁醇提取液,用氨试液洗涤2次,每次20ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液;另取黄芪对照药材1~2g,加水40ml,煎煮20分钟,滤过,滤液同法制成对照药材溶液;再取黄芪甲苷对照品,加甲醇制成每lml含2mg的溶液,作为对照品溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述对照品溶液、对照药材溶液、供试品溶液各10μl,分别点于同一硅胶g薄层板上,以比例为13:7:2三氯甲烷-甲醇-水5℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;置365nm紫外光灯下显检视,显相同颜色的荧光斑点;(3)取本品及缺甘草阴性样品各0.5g,加水20ml,超声20分钟,滤过,用乙醚振摇提取2次,每次10ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取2次,每次15ml,合并正丁醇液,用氨试液振摇提取2次,每次20ml,合并氨试液,用盐酸调ph值至3~4;再用乙酸乙酯振摇提取2次,每次20ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液及缺甘草阴性样品溶液;另取炙甘草饮片0.5g,加水40ml,煎煮20分钟,滤过,同法制成对照药材溶液;取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述供试品溶液及缺甘草阴性样品溶液、对照药材溶液、对照品溶液各5μl,分别点于同一硅胶g薄层板上,以比例为13:7:2的三氯甲烷-甲醇-水5℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及365nm紫外光灯下检视,供试品色谱中,在与对照品、对照药材色谱相应的位置上,显相同颜色的斑点或荧光斑点;(4)取本品及缺肉桂阴性冻干粉各0.5g,加水5ml使其溶解,用乙醚振摇提取2次,每次15ml,合并乙醚液,挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液;另取肉桂饮片0.5g,加水40ml,煎煮20分钟,滤过,同法制成对照药材溶液;再取肉桂酸对照品,用40%甲醇制成1ml含0.2mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述供试品溶液及缺肉桂阴性样品溶液、对照药材溶液、对照品溶液各3μl,点于同一硅胶gf254薄层板上,以比例为5:5:0.3环己烷-乙醚-冰醋酸为展开剂,展开,取出,晾干,置254nm紫外光灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点或荧光斑点;【检查】水分照(《中国药典》2015版四部通则0832)第二法测定不得过12.0%;【浸出物】照醇溶性浸出物测定法(《中国药典》2015版四部通则2201)项下热浸法测定,浸出物为18%~81.0%;【指纹图谱】照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;色谱柱:inertsustain-aq-c18,以乙腈为流动相a,以0.1%磷酸溶液为流动相b,流速为1ml/min,检测波长为200nm,具体梯度洗脱程序为:流动相梯度洗脱程序参照物溶液的制备取毛蕊异黄酮葡萄糖苷对照品适量,精密称定,加40~60%甲醇制成每1ml含毛蕊异黄酮葡萄糖苷50.4592μg/ml的溶液,即得;供试品溶液的制备取本品1.5g,精密称定,置50ml三角锥瓶中,加40%甲醇40ml超声30min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取2次,每次15ml,合并正丁醇溶液,用20ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加40~60%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得;测定法分别精密吸取参照物溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录70min的色谱图,即得;供试品指纹图谱中应分别呈现相应的参照物色谱峰保留时间相同的色谱峰;按中药色谱指纹图谱相似度评价系统计算,供试品指纹图谱与对照指纹图谱的相似度不得低于0.85,对照指纹图谱见图1。【含量测定】:人参照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为190nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000;按规定梯度洗脱程序进行梯度洗脱,梯度洗脱程序为:流动相梯度洗脱程序对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得;供试品溶液的制备取本品0.5g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取2次,每次10ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取2次,每次20ml,合并正丁醇液,用氨试液洗涤2次,每次20ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液10ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得;测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得;本品按干燥品计算,每1g含人参以人参皂苷rg1、rb1、re的总含量计,应为0.6mg/g~2.4mg/g;【含量测定】黄芪照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验十八烷基硅烷键合硅胶为填充剂;以乙腈-水(35:65)为流动相;蒸发光散射检测器检测;理论板数按黄芪甲苷峰计算应不低于4000;对照品溶液的制备取黄芪甲苷对照品适量,精密称定,加甲醇制成每lml含0.5mg的溶液,即得;供试品溶液的制备取本品0.5g,精密称定,精密加水40ml,称定重量,超声处理(功率720w,频率40khz)30分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取2次,每次20ml,合并正丁醇提取液,用氨试液洗涤2次,每次20ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。本品按干燥品计算,每1g含黄芪以黄芪甲苷(c41h68o14)计,应为0.10mg/g~1.40mg/g。实施例21检测方法【性状】本品为棕色至棕褐色粉末,气微香,味微苦甜;【鉴别】(1)取本品粉末1.5g,加三氯甲烷60ml,加热回流2小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理40分钟,吸取上清液加4倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液;另取人参对照药材1g,同法制成对照药材溶液;再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rf对照品及人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述供试品溶液、对照药材溶液、对照品溶液各1~2μ1,分别点于同一硅胶g薄层板上,以比例为15:40:22:10的三氯甲烷-乙酸乙酯-甲醇-水15℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和365nm紫外光灯下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的斑点,阴性色谱在对照色谱相应的位置上无斑点;(2)取本2g,加水35ml,超声处理40分钟,滤过,滤液用水饱和正丁醇振摇提取4次,每次30ml,合并正丁醇提取液,用氨试液洗涤4次,每次40ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液;另取黄芪对照药材1~2g,加水60ml,煎煮40分钟,滤过,滤液同法制成对照药材溶液;再取黄芪甲苷对照品,加甲醇制成每lml含2mg的溶液,作为对照品溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述对照品溶液、对照药材溶液、供试品溶液各10μl,分别点于同一硅胶g薄层板上,以比例为13:7:2三氯甲烷-甲醇-水15℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰;供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;置365nm紫外光灯下显检视,显相同颜色的荧光斑点;(3)取本品及缺甘草阴性样品各1.5g,加水60ml,超声40分钟,滤过,用乙醚振摇提取4次,每次30ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取4次,每次25ml,合并正丁醇液,用氨试液振摇提取4次,每次40ml,合并氨试液,用盐酸调ph值至3~4;再用乙酸乙酯振摇提取4次,每次40ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液及缺甘草阴性样品溶液;另取炙甘草饮片1.5g,加水60ml,煎煮40分钟,滤过,同法制成对照药材溶液;取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述供试品溶液及缺甘草阴性样品溶液、对照药材溶液、对照品溶液各10μl,分别点于同一硅胶g薄层板上,以比例为13:7:2的三氯甲烷-甲醇-水10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及365nm紫外光灯下检视,供试品色谱中,在与对照品、对照药材色谱相应的位置上,显相同颜色的斑点或荧光斑点;(4)取本品及缺肉桂阴性冻干粉各1.5g,加水15ml使其溶解,用乙醚振摇提取3次,每次35ml,合并乙醚液,挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液;另取肉桂饮片1.5g,加水60ml,煎煮40分钟,滤过,同法制成对照药材溶液;再取肉桂酸对照品,用65%甲醇制成1ml含0.2mg的溶液,作为对照品溶液;照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述供试品溶液及缺肉桂阴性样品溶液、对照药材溶液、对照品溶液各3μl,点于同一硅胶gf254薄层板上,以比例为5:5:0.3环己烷-乙醚-冰醋酸为展开剂,展开,取出,晾干,置254nm紫外光灯下检视,供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点或荧光斑点;【检查】水分照(《中国药典》2015版四部通则0832)第二法测定不得过12.0%;【浸出物】照醇溶性浸出物测定法(《中国药典》2015版四部通则2201)项下热浸法测定,浸出物为18%~81.0%;【指纹图谱】照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;色谱柱:inertsustain-aq-c18,以乙腈为流动相a,以0.1%磷酸溶液为流动相b,流速为1ml/min,检测波长为220nm,具体梯度洗脱程序为:流动相梯度洗脱程序参照物溶液的制备取毛蕊异黄酮葡萄糖苷对照品适量,精密称定,加60%甲醇制成每1ml含毛蕊异黄酮葡萄糖苷50.4592μg/ml的溶液,即得;供试品溶液的制备取本品3.5g,精密称定,置50ml三角锥瓶中,加60%甲醇60ml超声60min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取3次,每次35ml,合并正丁醇溶液,用40ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加60%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得;测定法分别精密吸取参照物溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录70min的色谱图,即得;供试品指纹图谱中应分别呈现相应的参照物色谱峰保留时间相同的色谱峰;按中药色谱指纹图谱相似度评价系统计算,供试品指纹图谱与对照指纹图谱的相似度不得低于0.85,对照指纹图谱见图1;【含量测定】:人参照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为210nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000;按规定梯度洗脱程序进行梯度洗脱,梯度洗脱程序为:流动相梯度洗脱程序对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得;供试品溶液的制备取本品1.5g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取4次,每次20ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取4次,每次40ml,合并正丁醇液,用氨试液洗涤4次,每次40ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液30ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得;测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得;本品按干燥品计算,每1g含人参以人参皂苷rg1、rb1、re的总含量计,应为0.6mg/g~2.4mg/g;【含量测定】黄芪照高效液相色谱法(《中国药典》2015年版四部通则0512)测定;色谱条件与系统适用性试验十八烷基硅烷键合硅胶为填充剂;以乙腈-水(35:65)为流动相;蒸发光散射检测器检测;理论板数按黄芪甲苷峰计算应不低于4000;对照品溶液的制备取黄芪甲苷对照品适量,精密称定,加甲醇制成每lml含0.5mg的溶液,即得;供试品溶液的制备取本品1.5g,精密称定,精密加水60ml,称定重量,超声处理(功率720w,频率40khz)60分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取6次,每次40ml,合并正丁醇提取液,用氨试液洗涤4次,每次40ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。本品按干燥品计算,每1g含黄芪以黄芪甲苷(c41h68o14)计,应为0.10mg/g~1.40mg/g。为了进一步验证本发明的有效性,发明人进行了以下试验,具体如下:1保元药物组合物物质基准研究1.1工艺描述和流程图1.1.1处方人参3.69g、黄芪7.38g、甘草1.85g、肉桂0.74g、生姜一片1.1.2物质基准的制法以上四味药,加生姜,将处方量放大20倍,即人参74g,黄芪148g,甘草37g,肉桂15g,生姜20片,按中药配方颗粒根茎类药材加水量标准,加水量为处方量的7倍,即1920ml,浸泡40min,煎煮1h,200目滤布过滤,滤液低温真空浓缩至与投料量比为1﹕3的浸膏,低温冷冻干燥,即得物质基准对应实物,置低温干燥处储存。1.1.4操作过程1.1.4.1煎煮次数及每煎投料量依据原文记载,煎煮次数为一次,煎煮投料量为日处方量的七倍。为保持与古籍记载工艺的一致性,确定煎煮次数为一次,每煎投料量为日处方量的七倍。1.1.4.2饮片前处理保元汤处方中含有两味药,分别为人参、甘草。方中人参饮片规格为2mm的薄片,黄芪饮片规格为2~5mm的厚片,甘草饮片规格为2~5mm的厚片,肉桂饮片。1.1.4..3煎煮过程(1)加热方式考察以提取液中的人参皂苷rg1、rb1、re总量、黄芪甲苷的提取总量及出膏率为考察指标,对明火和电子煎药罐两种加热方式进行单因素对比试验研究。从煎煮时间、出膏率、指标成分总量综合分析,两种加热方式存在差异。(2)煎煮时间考察由于处方原文中煎煮终点为提取液体积,而以提取液体积作为煎煮终点在煎煮过程中操作不便,同时以固定加热方式,故对煎煮时间代替提取液体积作为煎煮终点进行研究。1.1.4..4滤过、浓缩与干燥工艺考察对提取液采用常压滤过工艺考察,以药液澄明度为指标。结果:提取液采用200目滤布趁热过滤,滤液澄清,无杂质。确定滤过方式为:200目滤布常压趁热过滤。由于提取液体积约为1000ml,故对提取液采用常压浓缩和减压浓缩进行考察,以浓缩液中人参皂苷rg1、rb1、re总量,黄芪甲苷含量及转移率为考察指标。结果:含量无显著差异,减压浓缩略微比常压浓缩含量及转移率偏高一点。确定浓缩方式为:减压浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏。对浓缩液进行冷冻干燥,浓缩液经过冷冻干燥后,能够收集到疏松的干膏粉,冷冻干燥方式可行。1.1.4..5包装内包材真空密封袋,密封;1.1.4..6贮存置阴凉库保存。1.1.5工艺参数确定的依据根据经典名方目录中记载的方法:人参一钱、黄芪二钱、甘草五分,肉桂二分,右加生姜一片,水煎服。本研究采用电子煎药罐煎煮,以出膏率、人参皂苷rg1、rb1、re总量,黄芪甲苷含量、指纹图谱为指标,研究物质基准饮片前处理、加热方式、煎煮时间、滤过及干燥等工艺相关参数,从而确定保元汤物质基准工艺。1.1.5.1前处理保元汤处方中含有四味药,分别为人参、黄芪、甘草、肉桂,均为饮片规格,适合汤剂的煎煮。1.1.5.2煎煮过程工艺研究考察明火及电子煎药罐两种加热方式,结果从煎煮时间、出膏率、人参皂苷rg1、rb1、re总量,黄芪甲苷含量综合分析,两种加热方式差异不大,由于电子煎药罐更为便捷,故选择加热方式为电子煎药罐。对煎煮时间代替提取液体积作为煎煮终点进行研究,确定保元汤物质基准煎煮时间为1h。1.1.5.3滤过、浓缩、干燥过程确定固液分离采用200目滤布趁热滤过。提取液体积约为1000ml,减压浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浓缩液经过冷冻干燥(预冻温度:-20~-45℃,冷冻干燥温度-50~-70℃,真空度<300pa)后,能够收集到疏松的干膏粉,确定冷冻干燥方式可行。1.1.5.4工艺验证过程通过对三批物质基准相应批次的药材、饮片、提取液、物质基准取样,测定指标成分含量及指纹图谱,从人参皂苷rg1、rb1、re总量,黄芪甲苷含量上分析,三批验证提取液与物质基准数据均基本平行,且工艺过程中物质传递损失较小,从指纹图谱上分析,药材、饮片、提取液、物质基准色谱图中共有峰,其相对保留时间rsd小于2.0%。1.1.5.5包装内包材真空密封袋,密封。1.1.5.6贮存置阴凉库保存。1.1.6主要设备及型号表1设备型号1.2工艺研究1.2.1前处理根据古籍原文及考证结果,本处方所用饮片由药材按相应的炮制标准制得,人参饮片规格为2mm的薄片,黄芪饮片规格为2~5mm的厚片,甘草饮片规格为2~5mm的厚片,肉桂饮片,研究用饮片信息见表2。表2研究用饮片信息1.2.2煎煮1.2.2.1吸水性考察因人参药用部位为根,黄芪药用部位为根及根茎,甘草药用部位为根及根茎部分,肉桂药用部位为干燥树皮,都为极易吸水,故进行药材吸水性试验。实验方法:取人参饮片74g、黄芪饮片148、甘草饮片37g、肉桂饮片15g,混合均匀,加7倍量的水(即1920ml),定时过滤,称量并记录过滤药材的重量,计算不同时间混合药材的吸水重量。结果见表3:表3吸水性试验考察表试验结果表明:混合药材的浸泡时间在40min后吸水重量趋于稳定,故将保元汤药材的提取浸泡时间优选为40min。1.2.2.2加水量考察保元汤处方中未规定煎煮的加水量,故先对煎煮的加水量进行考察,加水量设计处方量的7倍、9倍、11倍量三个梯度进行考察。将处方放大二十倍,即分别取人参74g(批号:rs-fs-2018070201y),黄芪148g(批号:hq-lx-2017110801y),甘草37g(批号:gc-dx-2017110401y),肉桂15g(批号:rg-pn-2018090701y),分别按7倍加水量(1920ml)、9倍(2470ml)、11倍量(3020ml)进行实验,按煎煮加水量因素表进行考察试验,见表4。表4煎煮加水量因素表各因素的取值以人参皂苷rg1、rb1、re总含量、黄芪甲苷的含量、转移率;含固率作为考察指标,详细结果见表5表5煎煮加水量因素含量汇总表结论:由加水量因素考察可知,人参皂苷rg1、rb1、re总含量、黄芪甲苷含量及转移率差异不显著,含固率7倍加水量较好,又结合《医疗机构中药煎药室管理规范》,确定保元汤物质基准煎煮加水量优选为7倍量水,即1920ml。1.2.2.3加热方式考察古代煎煮方式为明火煎煮,现拟用电炉作为明火加热方式,同时考虑到电子煎药罐加热方式通过功率控制加热效率,其控制火力作用更稳定,故拟采用出膏率、人参皂苷rg1、rb1、re总量,黄芪甲苷含量为指标,考察明火与电子煎药罐加热方式,若两种加热方式结果无差异,则选择电子煎药罐作为加热方式。称取4份20倍日处方量饮片人参74g(批号:rs-fs-2018070201y),黄芪148g(批号:hq-lx-2017110801y),甘草37g(批号:gc-dx-017110401y),肉桂15g(批号:rg-pn-2018090701y),加水1920ml,置于电子煎药罐中,加盖煎煮,武火煎煮至沸,文火煎煮至1000ml,提取液用200目滤布滤过,记录提取液体积;明火和电子煎药罐(武火为2200w,文火为400w)煎煮均制备两份提取液,按照指标成分的测定方法,测定提取液中人参皂苷rg1、rb1、re总量,黄芪甲苷含量、出膏率,考察明火和电子煎药罐(武火为2200w,文火为400w)加热方式对指标性成分的影响。结果见表6。中间体(提取液)出膏率测定方法:精密量取提取液20ml,置已干燥至恒重的蒸发皿w1中,在水浴上蒸干后,于105℃干燥3小时,置干燥器中冷却30分钟,迅速精密称定重量w2,详细见表6。表6加热方式考察结果试验结果表明,明火及电子煎药罐两种加热方式,含量测定指标测定总量无显著性差异,但煎煮时间明火较电子煎药罐时间短,出膏率有差别,综合分析,两种加热方式存在差异较小,基于煎煮方便,选择优选电子煎药罐煎煮。1.2.2.4煎煮时间的考察保元汤出自《简明医彀》(明·孙志宏),“治元气虚弱,精神倦怠,肌肉柔慢,饮食少进,面青白,睡卧宁静,……及有杂证,皆属虚弱,宜服。”由人参、黄芪、甘草、肉桂四味药物组成。原文记载煎煮方法为:“人参一钱、黄芪二钱、甘草五分、肉桂二分,右加生姜一片,水煎服。”依据《历代度量衡》及《中国历代度、量、衡制演变简表》对保元汤中“制法及用法”中的数据单位同现代的数据单位进行换算。一钱为现今的3.69克,一分为0.37克,即保元汤煎煮工艺的“制法”可描述为取人参3.69g黄芪7.38g甘草1.85g肉桂0.74g,生姜1片,加水1920ml,浸泡30min,加热煎煮,滤弃药渣即得。根据经典名方目录中记载的方法,结合卫生部、国家中医药管理局《医疗机构中药煎药室管理规范》(国中医药发〔2009〕3号)进行煎煮制备“保元汤物质基准”,“保元汤物质基准”的制备应贴近传统、遵循古方原则。加热煎煮容器以电子煎药罐为宜,遵循古方的煎煮方法,已规定加水量及煎煮次数,故对煎煮时间进行考察。将原处方量放大20倍,分别取人参74g(批号:rs-fs-2018070201y),黄芪148g(批号:hq-lx-2017110801y),甘草37g(批号:gc-dx-2017110401y),肉桂15g(批号:rg-pn-2018090701y)、生姜20片,按煎煮时间因素表进行试验,见表7:表7煎煮时间因素表各因素的取值以人参皂苷rg1、rb1、re的总量和黄芪甲苷的转移率作为考察指标,详细结果见表8:表8煎煮时间转移率考察结论:综上提取液含量、转移率、含固率可知,煎煮1h药液中指标性成分的含量较高,转移率和含固率较好,故保元汤煎煮时间优选为1h。1.2.2.5煎煮工艺总结综上,暂定保元汤物质基准煎煮优选工艺为:取人参74g、黄芪148g、甘草37g、肉桂15g、生姜20片,加水1920ml,置于电子煎药罐中,加盖煎煮,武火煎煮至沸,文火煎煮1h,200目滤布滤过,即得。1.2.3滤过、浓缩与干燥1.2.3.1滤过方法考察为避免有效成分在工业生产中的损失,故对过滤方法进行考察。试验方法:分别取3份人参饮片(批号:rs-dh-2018070201y)74g、黄芪饮片(批号:hq-lf-2017110801y)148g、甘草饮片(批号:gc-hm-2017110601y)37g、肉桂饮片(批号:rg-rx-2018090701y)15g、生姜20片,加水1920ml,浸泡30min,加热煎煮1h,分别不过滤、2层纱布过滤、200目滤布过滤,分别计算3种药液的含固率,结果见表9:表9过滤实验结果结论:过滤的方法对药液澄明度无显著差异,但因原液和2层纱布过滤的溶液易沉淀,故“保元汤物质基准”的过滤方法优先选用200目滤布过滤。1.2.3.2浓缩方法考察为保证药液中的有效成分损失最小,参阅《中药提取液浓缩工艺和设备现状及问题分析》,结合环保节能降耗及企业的实际情况,故采用常压加热浓缩和真空低温浓缩两种方法进行考察。实验方法:取人参饮片(批号:rs-dh-2018070201y)222g、黄芪饮片(批号:hq-lf-2017110801y)444g、甘草饮片(批号:gc-hm-2017110601y)111g、肉桂饮片(批号:rg-rx-2018090701y)45g、生姜60片,加水5760ml,浸泡40min,加热煎煮1h,过滤,分别取提取液700ml采用常压和真空两种方法进行浓缩,浓缩至100ml,以便于下一步工序的操作。分别计算2种浓缩液的含固率、人参皂苷rg1、rb1、re的总含量及黄芪甲苷的含量,各因素的取值以人参皂苷rg1、rb1、re的总含量及黄芪甲苷的转移率作为考察指标,结果见表10:表10浓缩方法考察数据试验结果表明:常压浓缩与真空浓缩相比较,真空浓缩略微比常压浓缩含量及转移率偏高一点。两份浓缩液的含固率相差不大,而常压浓缩所需时间为4h,真空浓缩所需时间为2h,因考虑到浓缩时间及环保节能,故优选采用真空的方法进行浓缩,浓缩至药液为100ml左右。1.2.3.3浓缩真空度的考察在确定了浓缩方法为低温真空浓缩之后,真空度的高低是否会对有效成分产生影响,所以现对真空度进行考察,进一步确定浓缩工艺参数,结合环保节能降耗和企业实际情况,确保浓缩过程中有效成分损失最小,故采用减压旋蒸浓缩法对“保元汤物质基准”进行浓缩,对浓缩的真空度进行考察。实验方法:取浓缩方法试验的提取液,均分成4份,每份煎液700ml,分别采用真空度为-0.04mpa、-0.06mpa和-0.08mpa三种方法进行浓缩,浓缩至100ml,以便于下一步工序的操作。分别计算3种浓缩液的含固率、人参皂苷rg1、rb1、re的总含量及黄芪甲苷的含量,各因素的取值以人参皂苷rg1、rb1、re的总含量及黄芪甲苷转移率作为考察指标,结果见表11:表11浓缩真空度考察数据试验结果表明:真空度对含固率无显著影响,真空度-0.04mpa、-0.06mpa和-0.08mpa的浓缩所用时间分别为4.5h、3.5h和2h,真空度越大所用时间就越少。人参皂苷rg1、rb1、re的总量和转移率最大为-0.06mpa,黄芪甲苷的含量和转移率在-0.08mpa时含量和转移率为最大,且都比-0.04mpa的要高,因考虑到浓缩时间及环保节能和有效成分最大程度的保留,故采用真空度为-0.08mpa~-0.06mpa的方法进行浓缩。1.2.3.4浓缩温度的考察减压旋蒸浓缩法在浓缩过程中,浓缩温度的高低是否会对有效成分产生影响,对浓缩过程的耗能影响有多大都需要进一步进行考察,所以现对真空浓缩过程中的浓缩温度进行考察,进一步确定浓缩工艺参数,低温浓缩对有效成分的保留具有重要的意义,故本试验确定45℃、50℃、55℃和60℃为考察温度界限值。实验方法:分别取人参饮片(批号:rs-dh-2018070201y)222g、黄芪饮片(批号:hq-lf-2017110801y)444g、甘草饮片(批号:gc-hm-2017110601y)111g、肉桂饮片(批号:rg-rx-2018090701y)45g、生姜60片,加水5760ml,浸泡40min,加热煎煮1h,分别过滤,将所得滤液分别成4份,每份800ml,分别采用温度为45℃、50℃、55℃和60℃四种方法进行浓缩,浓缩至100ml,以便于下一步工序的操作。分别计算4种浓缩液的含固率及人参皂苷rg1、rb1、re的总量和黄芪甲苷的含量,各因素的取值以人参皂苷rg1、rb1、re的总量和黄芪甲苷的转移率作为考察指标,结果见表12:表12浓缩温度考察数据试验结果表明:温度对含固率无显著影响,温度为45℃、50℃、55℃和60℃的浓缩所用时间分别为4.5h、4h、3h和2h,温度越低所用时间越长。浓缩温度对人参皂苷rg1、rb1、re的总量和黄芪甲苷的含量也无显著影响。所以,在低温真空浓缩条件下时间越短越好,故采用温度优选为55℃~60℃的方法进行浓缩。1.2.3.5干燥取浓缩工艺考察液进行冷冻干燥,(预冻温度:-20~-50℃,冷冻干燥温度-45~-60℃,真空度<300pa),观察干燥情况,结果见表13。表13物质基准工艺考察结果综上所述:暂定保元汤物质基准制备工艺为:人参饮片74g、黄芪饮片148g、甘草饮片37g、肉桂饮片15g、生姜20片,加水1920ml,浸泡40min,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,浓缩真空度优选为-0.06mpa~-0.08mpa,浓缩温度优选为55℃~60℃,浓缩至药液浓缩至药液为100ml左右,经冷冻干燥,收集干膏粉,即得。1.2.3.6相关工艺参数根据“经典名方物质基准”制作的各工序步骤,将“保元汤物质基准”制备的相关质量控制参数汇总如下表14。表14质量控制参数“保元汤物质基准”的制备工艺参数、质量控制指标详见上表,保元汤大生产过程中的工艺参数与质量控制指标均参照以上数据进行调整。1.2.4关键工艺步骤和中间体1.2.4.1工艺验证样品制备平行称取3份饮片人参饮片(rs-th-2018070201y)74g、黄芪饮片(hq-lf-2017110801y)148g、甘草饮片(gc-ds-2017110801y)37g、肉桂饮片(rg-cx-2018090701y)15g、生姜20片,加水1920ml,置于电子煎药罐中,加盖煎煮,武火煎煮至沸,文火煎煮时间1h,200目滤布趁热滤过,滤液留样50ml,其余滤液减压浓缩,浓缩液留样50ml,其余浓缩液经冷冻干燥。(预冻温度:-20~-45℃,冷阱温度:-50~-70℃,真空度:<300pa),收集干膏粉,计算出膏率。取样,测定干膏粉中水分、浸出物、人参皂苷rg1、rb1、re的总量、黄芪甲苷含量,并分别计算工艺过程中含量转移率。另按照上述工艺,进行单味饮片及药材提取液制备,进行指纹图谱测定。8.2.4.2工艺验证样品测定结果见表15~16,图2:表15保元汤物质基准工艺验证检测结果表16保元汤物质基准工艺验证相对保留时间结果样品名称123456789101112甘草药材/1.101.23//2.29///3.47//黄芪药材1.0//1.491.772.35/2.80/3.496.026.27提取液11.01.091.221.491.782.352.552.803.403.485.976.23提取液21.01.091.221.501.782.352.552.813.413.485.986.23提取液31.01.091.221.491.782.352.552.803.413.495.986.23浓缩液11.01.091.231.511.802.382.582.843.463.536.066.32浓缩液21.01.091.231.511.802.382.582.843.463.536.066.31浓缩液31.01.081.221.501.802.382.572.863.513.596.236.49物质基准11.01.081.221.501.782.362.562.823.443.516.056.32物质基准21.01.081.221.501.792.372.572.843.443.526.086.34物质基准31.01.081.221.491.792.382.582.843.443.526.066.33平均值1.01.091.221.501.792.362.562.833.443.516.056.31rsd(%)00.450.420.440.561.160.560.720.961.021.271.251.2.4.3工艺确定从人参皂苷rg1、rb1、re的总量、黄芪甲苷总量上分析,三批验证提取液与干膏粉数据均基本平行,且工艺过程中物质传递损失较小;从浸出物、水分上分析,三批验证干膏粉无明显差异。从指纹图谱上分析,峰2、3、6、10为甘草色谱峰,峰1、4、5、8、11、12为黄芪色谱峰。暂以1号峰为参照峰计算相对保留时间,结果表明、药材、饮片、提取液、干膏粉色谱图中共有峰,其相对保留时间rsd小于2.0%,故所确定的物质基准制备工艺稳定可行。综上,确定物质基准优选工艺为:人参74g,黄芪148g,甘草37g,肉桂15g,生姜20片,置于9l电子煎药罐中,加水1920ml,加盖煎煮,武火煮沸转为文火煎煮保持微沸,煎煮1h,200目滤布趁热滤过,滤液在真空度为-0.08mpa~-0.06mpa、温度55℃~60℃下真空浓缩,浓缩至药液与饮片总投料量比为1﹕3的浸膏,浸膏在预冻温度:-20~-45℃、冷冻干燥温度-50~-70℃、真空度<300pa条件下冷冻干燥,收集干膏粉,即得。1.2.4.4多批次物质基准的制备目前饮片以5个产地,不少于15个批次为原则进行收集;处方中人参收集黑龙江伊春市铁力市、吉林省敦化市、吉林省通化市、吉林省白山市抚松县、吉林省白山市靖宇县5个产地,黄芪收集河北保承市、内蒙古固阳县、甘肃渭源县莲峰镇、甘肃陇西县、甘肃华亭县河西乡5个产地,甘草收集甘肃定西、甘肃兰州、甘肃张掖、新疆哈密和内蒙东胜5个产地,肉桂收集广西平南县、广东省高要市、广西防城港市、广西岑溪市、广西容县5个产地,每个产地3批,制备15批次物质基准。15批饮片排序组合结果见下表17。注:编号1~15,即为15个组合方式按照确定工艺,采用20倍日处方量进行投料,提取、浓缩、冷冻干燥。得到15批物质基准对应实物,15批次物质基准出膏率测定结果见下表17,测定范围为9.85%~17.52%,平均值为13.97%,sd值2.44,,±3sd值范围为6.65~21.28%,故确定物质基准出膏率范围为6.5%~21.5%。表17多批次物质基准批次信息1.3关键质量属性的研究(1)保元汤物质基准关键质量属性研究①定量研究指标的筛选含量测定指标的选择,结合处方功效主治及处方药味在处方中的位置,综合确定含量测定指标。由于工艺研究确定水为溶剂,故保元汤物质基准中水溶性成分较多,其中君药人参、黄芪主要成分有皂苷类、三萜类、黄酮类,故测定君药人参皂苷类成分人参皂苷rg1、rb1、re的总量;黄芪甲苷的含量,以综合评价物质基准质量。②含量测定研究a指标成分定量研究检测方法详见“保元汤物质基准”质量标准正文。三批验证检测结果见表18:表18工艺验证测定结果b定量指标检测结果汇总表表19工艺验证测定结果(2)关键质量属性的影响因素及控制办法①饮片质量药材采集固定选定的产地、基原,各味药材严格按照确定炮制工艺参数进行炮制,确保饮片的质量。②物质基准工艺过程控制a煎煮采用电子煎药罐为煎煮器具,电加热方式,武火煮沸、文火煎煮,对火候、煎煮时间、滤过材质等工艺参数进行控制。b冷冻干燥干燥方式为冷冻干燥,控制干膏水分小于12.0%。c包装、贮存物质基准包装包材为真空包装袋,密封,阴凉库保存。8.3.2药材、饮片、中间体与对应实物的相关性研究以关键质量属性(人参皂苷rg1、rb1、re的总量、黄芪甲苷总量和指纹图谱)为指标,进行药材、饮片、中间体与对应实物的相关性研究。8.3.2.1测定方法(1)含量测定方法对应实物(干膏粉)人参皂苷rg1、rb1、re的总量、黄芪甲苷测定方法详见“保元汤物质基准质量标准正文”。中间体供试品制备方法如下:①人参皂苷rg1、rb1、re的总量提取液:精密量取提取液25ml,置分液漏斗中,加氯仿萃取3次,每次15ml,弃去氯仿层,水层用水饱和的正丁醇溶液萃取3次,每次30ml,合并正丁醇层,用氨试液洗涤2次,每次30ml,弃去按试液层,正丁醇层用20ml正丁醇饱和水洗涤1次,弃去水层,正丁醇层蒸干,残渣加甲醇溶解并转移至5ml容量瓶中,加甲醇定容至刻度,摇匀,滤过,取续滤液,即得供试品溶液。②黄芪甲苷提取液:精密量取提取液25ml,置50ml三角锥瓶中,超声处理(功率240w,频率45khz)30分钟,用水饱和正丁醇萃取4次,每次25ml,合并正丁醇层,用氨试液振摇提取3次,每次30ml,弃去氨试液层,收集正丁醇液蒸干,残渣加甲醇溶解,转移至5ml容量瓶中,加甲醇定容至刻度,摇匀,滤过,取续滤液,即得供试品溶液。(2)指纹图谱测定方法药材、饮片、中间体(提取液)、对应实物(干膏粉)测定方法,根据15批次物质基准指纹图谱测定结果,标出10个共有峰,并生成对照指纹图谱,计算相似度。药材、饮片、中间体供试品制备方法如下:a甘草药材溶液:按处方称取甘草药材粉末(过四号筛)2.5g,精密称定,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。b甘草饮片溶液:按处方称取甘草饮片粉末(过四号筛)2.5g,精密称定,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。c人参饮片溶液:按处方称取人参饮片粉末(过四号筛)2.5g,精密称定,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。d中间体(提取液):精密量取提取液5ml,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。e中间体(浓缩液):精密量取浓缩液2ml,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。1.3.2.2相关性研究(1)多批次相关性研究①含量测定多批次含量测定结果见表20~表21。多批次黄芪甲苷、人参皂苷rg1、rb1、re的总量、提取总量,离散度均较小,相关性良好。表20不同批次黄芪甲苷含量测定结果表21不同批次人参皂苷rg1、rb1、re的总量含量测定结果②指纹图谱黄酮类成分指纹图谱:多批次指纹图谱相对保留时间结果见表22,15批样品共有峰相对保留时间rsd小于3.0%;采用中药色谱指纹图谱相似度评价软件系统(2012版),以共有峰对15批样品与对照指纹图谱的相似度进行计算,按指纹图谱计算的相似度,均大于0.85,说明相关性良好。表22相对保留时间编号12345678910111211.001.081.211.471.752.352.532.803.353.455.886.1321.001.081.211.471.752.352.542.813.383.475.986.2331.001.081.211.481.772.392.582.863.443.536.056.3041.001.081.211.481.762.362.542.813.393.496.026.2851.001.081.211.481.772.362.552.833.413.516.056.3161.001.081.211.481.762.362.552.833.423.516.096.3571.001.081.211.491.782.372.552.833.433.526.116.3881.001.081.211.491.782.372.562.843.433.536.136.4091.001.091.221.491.782.372.562.843.443.526.106.36101.001.091.221.491.772.362.552.823.413.506.016.27111.001.081.221.491.772.362.562.823.413.506.036.29121.001.091.221.491.772.372.562.833.433.526.066.32131.001.091.221.491.772.372.562.833.433.516.076.33141.001.081.221.491.772.372.562.833.433.526.086.35151.001.081.221.491.782.382.572.853.453.536.116.37平均值1.001.081.211.481.772.372.562.833.423.516.056.31rsd(%)00.240.320.450.500.450.530.550.750.661.051.09表23十五批物质基准相似度评价结果r对照图谱s2~s16依次为:byt-2019091001d、byt-2019091002d、byt-2019091003d、byt-2019091604d、byt-2019091605d、byt-2019091606dbyt-2019091807d、byt-2019091808d、byt-2019091809d、byt-2019092310d、byt-2019092311d、byt-2019092312d、byt-2019092413d、byt-2019092414d、byt-2019092415d(2)同批次药材、饮片、中间体、对应实物相关性研究①物质基准的制备取人参饮片(rs-th-2018070201y)74g、黄芪饮片(hq-lf-2017110801y)148g、甘草饮片(gc-ds-2017110801y)37g,肉桂(rg-cx-2018090701y)15g按确定工艺制备保元汤物质基准,提取液留样50ml,备用。②相关性研究测定结果a含量测定结果表24人参饮片-提取液-物质基准含量测定结果汇总表25黄芪饮片-提取液-物质基准含量测定结果汇总b指纹图谱测定结果见图3表26相对保留时间计算结果汇总注:药材、饮片经过水煮后同对照药材制备方式制备。结论:由以上结果可知,饮片、提取液、浓缩液、物质基准的色谱图的在相应位置均有相同的色谱峰,相对保留时间rsd%<2.0%,表明饮片到物质基准主要化学成分的一致性,符合质量标准的要求,说明制备工艺稳定。③相关性测定结果小结通过对饮片-提取液-浓缩液-物质基准中人参皂苷rg1、rb1、re的总量、黄芪甲苷含量测定,结果表明,人参皂苷rg1、rb1、re的总量、黄芪甲苷总量传递与工艺验证过程中传递结果一致。通过对饮片-提取液-浓缩液-物质基准过程中指纹图谱测定,结果表明物质基准中标出的12个共有峰传递良好,各峰相对保留时间rsd<2.0%,相关性良好。由上述测定结果确定,从饮片-提取液-浓缩液-物质基准研究过程中,指纹图谱、含量测定、出膏率的传递,呈现一定规律性,且相关性良好。1.3.3分析方法研究通过文献调研,我们汇总出保元汤物质基准中系统的化学成分及关键质量属性,并对建立物质基准关键质量属性的分析及验证方法;物质基准质量标准中各检测项目总结见表27。1.3.3.1分析方法1.3.3.2分析方法的验证列入标准项目的分析方法学验证,按照现行版《中国药典》中有关的指导原则执行,方法学验证资料如下:1.3.3.2.1性状通过对15批对应实物(干膏粉)的实际性状观察,记录,结果见表28,图4:表28性状鉴别结果结论:根据15批次对应实物(干膏粉)样品的实际观察情况,暂定本品为棕色至棕褐色粉末,气微香,味微苦甜。1.3.3.2.2鉴别本品由四味中药组成,系用水煎煮工艺制成的复方制剂,选用了专属性强,快速、简便、重现性好的薄层鉴别方法,对方中人参、黄芪、甘草、肉桂的主要特征成分进行鉴别试验研究。结果概述如下:①人参人参主要成分为皂苷类、糖类、挥发性成分、有机酸及其酯、甾醇及其苷、黄酮类、木质素、无机元素及维生素类等。主要含有人参皂苷,根据以上化学成分对人参进行如下薄层摸索实验。a建立薄层色谱方法参照《中国药典》2015版一部人参【鉴别】项进行方法摸索[58]取本品粉末1g,加三氯甲烷40ml,加热回流1小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理30分钟,吸取上清液加3倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取人参对照药材1g,加适量水煎煮30min,同法制成对照药材溶液。再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各5μ1,分别点于同一硅胶g薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和紫外光灯(365nm)下检视:结果分析:供试品色谱中,在与对照药材、对照品相应的位置上,显相同的斑点,且阴性无干扰。以人参皂苷rg1、rb1、re为对照品,以人参对照药材溶液作为对照,在紫外光下检视,方法可行,但供试品、对照品及对照药材溶液斑点较浅,故下一步对供试品、对照品及对照药材溶液点样量进行摸索。方法二:点样量摸索吸取“方法一”制备的三种样品溶液,将人参皂苷rg1、rb1、re对照品溶液、人参饮片溶液的点样量增加至10μl,阴性样品的点样量为5μl,供试品点样量变为5μl,8μl和10μl,分别点于同一硅胶g薄层板上,三氯甲烷-乙酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和紫外光灯(365nm)下检视。结果表明,人参皂苷rg1、rb1、re对照品溶液与人参饮片点样量均为10μl,供试品溶液点样量为10μl,相应位置下的显色斑点均清晰可见,没有出现拖尾现象,故确定人参皂苷rg1、rb1、re的对照品浓度为1mg/ml点样量10μl,人参对照药材溶液0.5g/ml点样量10μl,供试品溶液点样量10μl。综合分析,确定薄层鉴别方法为:取本品粉末1g,加三氯甲烷40ml,加热回流1小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理30分钟,吸取上清液加3倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取人参对照药材1g,加适量水煎煮30min,同法制成对照药材溶液。再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rf对照品及人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各1~2μ1,分别点于同一硅胶g薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和紫外光灯(365nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的斑点,阴性色谱在对照色谱相应的位置上无斑点。b耐用性考察按上述确定方法进行耐用性实验,考察自制板与机制板(青岛海洋化工厂),不同展开温度及相对湿度的影响。结论:通过对低温低湿、常温、高温高湿环境及不同厂家的硅胶g薄层板进行耐用性考察,结果不同影响因素下,此人参薄层鉴别条件耐用性良好。故将此薄层鉴别方法列入正文c方法确定取本品粉末1g,加三氯甲烷40ml,加热回流1小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理30分钟,吸取上清液加3倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取人参对照药材1g,加适量水煎煮30min,同法制成对照药材溶液。再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rf对照品及人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各1~2μ1,分别点于同一硅胶g薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和紫外光灯(365nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的蓝紫色斑点,阴性色谱在对照色谱相应的位置上无斑点。d不同批次对应实物(干膏粉)薄层鉴别分别称取不同批次的对应实物(干膏粉)1g,同上述供试品制备方法制成供试品溶液。进行薄层鉴别。结果:不同批次物质基准的供试品色谱中,在与对照品色谱相应的位置上,显相同的蓝紫色斑点,阴性色谱在对照色谱相应的位置上无斑点。②黄芪黄芪主要化学成分为皂苷类和黄酮类,除此之外还含有多糖等。根据以上化学成分对黄芪进行如下薄层色谱的摸索实验。a建立薄层色谱方法方法一:参考《中国药典》2015年版一部“黄芪颗粒”项下薄层鉴别方法进行摸索取本品及缺黄芪的阴性样品各1.5g,取本品粉末1.5g,加水25ml,超声处理30分钟,滤过,滤液用水饱和正丁醇振摇提取2次,每次25ml,合并正丁醇提取液,用氨试液洗涤2次,每次30ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液。另取黄芪饮片1.5g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。再取黄芪甲苷对照品适量,加甲醇制成每1ml含2mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述四种溶液各5μl,点于同一硅胶g薄层板上,三氯甲烷_甲醇-水(13:7:2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。结果分析:供试品色谱中,在与黄芪甲苷、黄芪药材色谱相应的位置上,有相同的斑点,且阴性无干扰。以黄芪甲苷为对照品,以黄芪对照药材溶液作为对照,在紫外光(365nm)下检视,方法可行,斑点较供试品溶液浅,为了让斑点更加的清晰明了,故进一步对对照品及黄芪饮片溶液点样量进行摸索。方法二:点样量摸索吸取“方法一”制备的样品溶液(byt-2019091807d),黄芪甲苷对照品溶液、黄芪对照药材溶液、阴性样品的点样量不变10μl,供试品点样量变为5μl,8μl和10μl,分别点于同一硅胶g薄层板上,以三氯甲烷_甲醇-水(13:7:2)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;置紫外光灯(365nm)下显检视。结果表明,黄芪甲苷对照品溶液与黄芪饮片点样量均为10μl,供试品溶液点样量分别为5μl,8μl和10μl,相应位置下的显色斑点均清晰可见,没有出现拖尾现象,故确定黄芪甲苷对照品浓度为2mg/ml点样量10μl,黄芪饮片溶液1.5g/ml点样量10μl,供试品溶液点样量10μl。综合分析,确认薄层鉴别方法:取本品1.5g,加水25ml使溶解,超声处理30分钟,滤过,滤液用水饱和正丁醇振摇提取2次,每次25ml,合并正丁醇提取液,用氨试液洗涤2次,每次30ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液。另取黄芪对照药材1.5g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。再取黄芪甲苷对照品适量,加甲醇制成每1ml含2mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取上述四种溶液各5μl,点于同一硅胶g薄层板上,以三氯甲烷_甲醇-水(13:7:2)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外光灯(365nm)下检视。b耐用性考察按上述确定方法进行耐用性试验,考察自制硅胶g板与机制硅胶g板的薄层板(青岛海洋化工厂)以及不同展开高温高湿(温度42℃、湿度75%),以及相对低温低湿(温度4℃、湿度45%)等影响因素。结论:通过对低温、高温、常温、低湿、高湿环境及不同的硅胶g薄层板进行耐用性考察,结果不同影响因素下,此黄芪薄层鉴别条件耐用性良好。故将此薄层方法列入正文。c方法确定取本品1.5g,加水25ml,超声处理30分钟,滤过,滤液用水饱和正丁醇振摇提取2次,每次25ml,合并正丁醇提取液,用氨试液洗涤2次,每次30ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液。另取黄芪对照药材1.5g,加水50ml,煎煮30分钟,滤过,滤液同法制成对照药材溶液。再取黄芪甲苷对照品,加甲醇制成每lml含2mg的溶液,作为对照品溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取对照品溶液、对照药材溶液、供试品溶液各10μl,分别点于同一硅胶g薄层板上,以三氯甲烷_甲醇-水(13:7:2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;置紫外光灯(365nm)下显检视,显相同颜色的荧光斑点。d不同批次对应实物(干膏粉)薄层鉴别分别称取不同批次的对应实物(干膏粉)1.5g,同上述供试品制备方法制成供试品溶液。进行薄层鉴别。结论:不同批次物质基准的供试品色谱中,在与对照药材色谱和对照品色谱相应位置上,显相同颜色斑点。③甘草甘草主要化学成分为三萜类和黄酮类,除此之外还含有多糖、有机酸和香豆素等。根据以上化学成分对甘草进行如下薄层色谱的摸索实验a建立薄层色谱方法方法一:参考《中国药典》2015年版一部“甘草”项下薄层鉴别方法取对应实物(冻干粉)及缺甘草阴性冻干粉1g,加水40ml,超声30分钟,滤过,用乙醚振摇提取2次,每次10ml,弃去乙醚液,水液用水饱和正丁醇振摇提取3次,每次20ml,合并正丁醇液,用氨试液振摇提取2次,每次30ml,合并氨试液,用盐酸调ph值至3~4。再用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取炙甘草饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液。照薄层色谱法2015版四部(通则0502)试验,吸取上述四种溶液各5μl,分别点于同一用1%氢氧化钠溶液制备的硅胶g薄层板上,以三氯甲烷-甲醇-水(13:7:2)10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及紫外光灯(365nm)下检视。结论:供试品色谱中,在与甘草对照药材、对照品色谱相应的位置上,显示相同颜色的斑点,但斑点显色不清晰,分离效果较差,5μl的点样量较小,定量较难且不准确,故进行点样量摸索。方法二:点样量摸索吸取“方法一”制备的样品溶液(byt-2019091001d),黄芪甲苷对照品溶液、黄芪饮片溶液、阴性样品的点样量不变10μl,供试品点样量变为5μl,8μl和10μl,分别点于同一用1%氢氧化钠溶液制备的硅胶g薄层板上,以三氯甲烷-甲醇-水(13:7:2)5~10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及紫外光灯(365nm)下检视。结果分析:供试品色谱中,在与甘草对照药材和对照品色谱相应的位置上,显相同颜色的斑点,无斑点拖尾严重,故确定供试品溶液和对照品、对照药材溶液的点样量均为10μl。综合分析确定薄层鉴别方法:取对应实物(冻干粉)及缺甘草阴性冻干粉1g,加水40ml,超声30分钟,滤过,用乙醚振摇提取2次,每次10ml,弃去乙醚液,水液用水饱和正丁醇振摇提取3次,每次20ml,合并正丁醇液,用氨试液振摇提取2次,每次30ml,合并氨试液,用盐酸调ph值至3~4。再用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取炙甘草饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液。照薄层色谱法2015版四部(通则0502)试验,吸取上述三种溶液各10μl,分别点于同一用1%氢氧化钠溶液制备的硅胶g薄层板上,以三氯甲烷-甲醇-水(13:7:2)5~10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及紫外光灯(365nm)下检视。b耐用性考察按上述确定方法进行耐用性试验,考察自制板与机制板(青岛海洋化工厂)以及不同展开温湿度等影响因素。结论:通过对低温低湿、常温、高温环境进行耐用性考察,结果不同影响因素下,此薄层鉴别条件耐用性良好。c方法确定取对应实物(冻干粉)及缺甘草阴性样品1g,加水40ml,超声30分钟,滤过,用乙醚振摇提取2次,每次20ml,弃去乙醚液,水液用水饱和的正丁醇振摇提取3次,每次20ml,合并正丁醇液,用氨试液振摇提取2次,每次30ml,合并氨试液,用盐酸调ph值至3~4。再用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取(炙)甘草饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各10μl,分别点于同一硅胶g薄层板上,以三氯甲烷-甲醇-水(13:7:2)5~10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及紫外光灯(365nm)下检视,供试品色谱中,在与对照品、对照药材色谱相应的位置上,显相同颜色的斑点或荧光斑点。d不同批次对应实物(冻干粉)薄层鉴别分别称取不同批次的对应实物(冻干粉)1g,同上述供试品制备方法制成供试品溶液。进行薄层鉴别。结论:不同批次物质基准的供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点或荧光斑点。④肉桂肉桂主要含有桂皮醛、肉桂酸、二萜及其糖苷、黄烷醇及其多聚体,除此之外还含有黄酮类、多酚类等。根据以上化学成分对肉桂进行如下薄层色谱的摸索实验。a建立薄层色谱方法方法一:参考文献“五苓片中桂枝、茯苓的薄层色谱鉴别”进行摸索取对应实物(冻干粉)及缺肉桂阴性冻干粉各1g,加水10ml使其溶解,用乙醚振摇提取1次,每次25ml,合并乙醚液,挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液。另取肉桂饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。再取肉桂酸对照品,用50%甲醇制成1ml含0.2mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述四种溶液各3μl,点于同一硅胶gf254薄层板上,以环己烷-乙醚-冰醋酸(5:5:0.3)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。结论:供试品色谱中,在与肉桂对照药材、对照品色谱相应的位置上,显示相同颜色的斑点,但斑点显色不清晰,分离效果较差,5μl的点样量较小,定量较难且不准确,故进行点样量摸索。方法二:点样量摸索吸取上述四种溶液各10μl,分别点于同一硅胶gf254薄层板上,以环己烷-乙醚-冰醋酸(5:5:0.3)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。结果分析:供试品色谱中,在与肉桂对照药材和对照品色谱相应的位置上,显相同颜色的斑点,无斑点拖尾严重,故确定供试品溶液和对照品、对照药材溶液的点样量均为10μl。综合分析确定薄层鉴别方法:取对应实物(冻干粉)及缺肉桂阴性冻干粉各1g,加水10ml使其溶解,用乙醚振摇提取1次,每次25ml,合并乙醚液,挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液。另取肉桂饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。再取肉桂酸对照品,用50%甲醇制成1ml含0.2mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述四种溶液各3μl,点于同一硅胶gf254薄层板上,以环己烷-乙醚-冰醋酸(5:5:0.3)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视。b耐用性考察按上述确定方法进行耐用性试验,考察自制板与机制板(青岛海洋化工厂)以及不同展开温湿度等影响因素。结论:通过对低温低湿、常温、高温环境进行耐用性考察,结果不同影响因素下,此薄层鉴别条件耐用性良好。c方法确定取对应实物(冻干粉)及缺肉桂阴性冻干粉各1g,加水10ml使其溶解,用乙醚振摇提取1次,每次25ml,合并乙醚液,挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液。另取肉桂饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。再取肉桂酸对照品,用50%甲醇制成1ml含0.2mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述四种溶液各3μl,点于同一硅胶gf254薄层板上,以环己烷-乙醚-冰醋酸(5:5:0.3)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视d不同批次对应实物(冻干粉)薄层鉴别分别称取不同批次的对应实物(冻干粉)1g,同上述供试品制备方法制成供试品溶液。进行薄层鉴别。结论:不同批次物质基准的供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点或荧光斑点。8.3.3.2.3检查①水分照水分测定法(《中国药典》2015年版四部通则0832第二法),测定15批次物质基准水分,结果见表29。表29水分检查结果结果表明:15批物质基准水分最小值为6.3%,最大值为11.8%,平均值为8.7%,故暂定干膏水分为不得过12.0%。②浸出物中药材中的化学成分或大类成分多数可以在水或醇中溶出,进而进行浸出物测定。由于保元汤物质基准是水提取,选择水为溶剂建立浸出物测定意义不大,难以反映内在质量,故选溶剂时,要考虑大类成分的溶解度,选择乙醇进行浸出物测定,以控制物质基准的质量照浸出物测定法(《中国药典》2015年版四部通则2201醇溶性热浸法)测定:取本品约2g,精密称定,置100ml的锥形瓶中,精密加乙醇50ml,密塞,称定重量,静置1小时后,连接回流冷凝管,加热至沸腾,并保持微沸1小时。放冷后,取下锥形瓶,密塞,再称定重量,用乙醇补足减失的重量,摇匀,用干燥滤器滤过,精密量取滤液25ml,置已干燥至恒重的蒸发皿中,在水浴上蒸干后,于105℃干燥3小时,置干燥器中冷却30分钟,迅速精密称定重量。计算供试品中醇溶性浸出物的含量(%)。测定15批经典名方物质基准中浸出物的含量,结果见表30。表30不同批次物质基准浸出物测定结果结果表明:经不同批次饮片制成的15批物质基准浸出物最小值为27.8%,最大值为70.1%,平均值为49.5%,平均值的±3sd值为18.4%~80.5%。考虑到实际生产过程中的各种影响因素等,暂定本品醇溶性浸出物的含量为18.0%~81.0%。1.3.3.2.4指纹图谱由于单纯的指标成分的定性鉴别和定量分析,难以反映物质基准质量的优劣。作为衡量物质基准对应实物是否与物质基准基本一致的标准参照物,其质量应加强专属性鉴别和多成分、整体质量控制。因而物质基准质量控制要重视以指纹图谱为主的整体质量控制。①仪器与试药液相色谱仪:赛默飞-u3000色谱柱:inertsustain-aq-c18(250mm×4.6,5μm)试剂:乙腈为色谱纯;甲醇为色谱纯,水为超纯水;其他试剂均为分析纯。样品:15批“保元汤物质基准”对照品:甘草酸铵(批号:110731-201720)、毛蕊异黄酮葡萄糖苷(批号:111920-201606),均购自中国食品药品检定研究院②色谱条件的摸索方法一:参照文献“黄芪药材的hplc-uv指纹图谱研究”色谱条件与系统适用性试验inertsustain-aq-c18为色谱柱;以乙腈为流动相a,以0.1%磷酸溶液为流动相b,按下表进行梯度洗脱;流速为1ml/min,柱温25℃,检测波长为,210nm,流动相梯度洗脱见表31。表31流动相梯度洗脱表参照物溶液的制备取毛蕊异黄酮葡萄糖苷、甘草酸铵对照品适量,精密称定,加50%甲醇制成每1ml含毛蕊异黄酮葡萄糖苷、甘草酸的溶液,供试品溶液的制备取本品约2.5g,精密称定,置50ml三角锥瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得。测定法分别精密吸取参照物溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录60min的色谱图,即得,见图5。方法二:参考文献“黄芪药材的hplc-uv指纹图谱研究”更改流动相梯度,其他条件不变,具体梯度如下表32,见图6。表32流动相梯度洗脱表方法三:参考文献“黄芪药材的hplc-uv指纹图谱研究”更改流动相梯度,其他条件不变,具体梯度如下表33。表33流动相梯度洗脱表方法四:参考文献“黄芪药材的hplc-uv指纹图谱研究”更改流动相梯度,其他条件不变,具体梯度如下表34。表34流动相梯度洗脱表方法五:参考文献“黄芪药材的hplc-uv指纹图谱研究”更改流动相梯度,其他条件不变,具体梯度如下表35。表35流动相梯度洗脱表结果分析:方法一、二、三、四流动相梯度洗脱,色谱图效果分离效果均不理想,在方法五梯度条件下,色谱峰之间的分离度良好,因此选择该梯度下流动相体系。③供试品溶液制备方法的选择参照文献“黄芪药材的hplc-uv指纹图谱研究”a、50%甲醇溶液:取保元汤物质基准约2.5g,精密称定,置于锥形瓶中,精密加入50%甲醇50ml,超声处理(100w,40khz)45min,过滤,取续滤液,用0.45μm微孔滤膜滤过,即得。b、水饱和的正丁醇萃取:取本品约2.5g,精密称定,置50ml三角锥瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得。c、乙酸乙酯萃取:取本品约2.5g,精密称定,置50ml三角锥瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用乙酸乙酯溶液萃取2次,每次25ml,合并乙酸乙酯溶液,用30ml的氨试液萃取1次,弃去氨试液,乙酸乙酯层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得。按照上述色谱条件,记录同一色谱条件下不同提取方法色谱图,结果见图10。结果表明:从上述色谱图可知,第一和第三两个方法检测色谱峰分离效果均不理想,方法二各峰之间的分离效果较好,因此选择方法二作为供试品的制备方法。④峰归属及参照物的选择a药味归属按照物质基准制备方法,制备单味药材的冻干粉及缺单味药的阴性样品冻干粉,按照确定的指纹图谱供试品溶液制备方法制备供试品溶液,按照确定的色谱条件进行测定,确定冻干粉指纹图谱中各个色谱峰归属于哪味饮片,见图11和表36。表36药味归属表实验结论:物质基准色谱峰主要为黄芪及甘草特征峰,流动相空白无干扰。b参照物的选择其中毛蕊异黄酮葡萄糖苷色谱峰保留时间适中,响应值较高,达到基线分离,故选择毛蕊异黄酮葡萄糖苷作为对照品参照峰,并将对毛蕊异黄酮葡萄糖苷标记为峰s,同时将毛蕊异黄酮葡萄糖苷对照品作为参照物。⑤方法的初步确定色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;色谱柱:inertsustain-aq-c18,以乙腈为流动相a,以0.1%磷酸溶液为流动相b,流速为1ml/min,检测波长为210nm,具体梯度见下表37表37流动相梯度洗脱表参照物溶液的制备取毛蕊异黄酮葡萄糖苷对照品适量,精密称定,加50%甲醇制成每1ml含毛蕊异黄酮葡萄糖苷50.4592μg/ml的溶液,即得。供试品溶液的制备取本品约2.5g,精密称定,置50ml三角锥瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得。测定法分别精密吸取参照物溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录70min的色谱图,即得。⑥方法学考察a专属性及整体性试验为考察空白溶剂是否有干扰,结果空白溶剂无干扰。同时也对供试品整体性进行了考察,结果基本满足了信息量最大的原则(见表38,图12)表38流动相梯度洗脱表结果分析:通过空白溶剂图谱和供试品溶液色谱峰的比较发现空白无干扰。b精密度试验取本品约(批号:byt-2019091604d)2.5g,精密称定,按正文供试品溶液的制备和测定法项下操作,连续进样6次,对其相对保留时间及相对峰面积进行计算,其相对保留时间及相对峰面积具体测定结果见表39~表40。表39指纹图谱精密度相对保留时间计算结果表40指纹图谱精密度相对峰面积汇总结果c稳定性试验取本品约(批号:byt-2019091604d)2.5g,精密称定,按正文供试品溶液的制备和测定法项下操作,分别在制备后放置0、3、6、9、12、24小时进样测定,对其相对保留时间及相对峰面积进行计算,其相对保留时间及相对峰面积具体测定结果如下表41~表42,图14。表41指纹图谱稳定性相对保留时间计算结果表42指纹图谱稳定性相对峰面积汇总结果结果表明,相对保留时间差异不大,rsd<2.0%;相对峰面积rsd<12.0%。说明供试品溶液中在24小时内稳定性良好。d重复性实验取本品(批号:byt-2019091604d)约2.5g,(共6份),精密称定,按正文供试品溶液制备和测定法项下进行操作。对其相对保留时间及相对峰面积进行计算,其相对保留时间及相对峰面积具体测定结果如下表43~表44,图15。表43指纹图谱重复性相对保留时间计算结果表44指纹图谱重复性相对峰面积汇总结果结果表明,相对保留时间差异不大,rsd<2.0%;相对峰面积rsd<10.0%,说明本方法的重复性良好。⑦指纹图谱的建立a共有峰的标定采用正文【指纹图谱】项下的方法对15批供试品色谱峰进行测定,并对所得指纹图谱进行分析,选择15批样品指纹图谱中稳定性较好、响应值适宜的色谱峰为共有峰,共标定了12个共有峰。结果见图16。b对照图谱的建立采用国家药典委员会颁布的《中药色谱指纹图谱相似度评价软件系统2012年版》,根据15批次物质基准指纹图谱测定结果,以标出的12个共有峰进行校正,生成如下对照指纹图谱r。结果见图1。c相似度的计算采用中药色谱指纹图谱相似度评价软件系统,以共有峰对15批样品与对照指纹图谱的相似度进行计算,结果见表45。表45十五批物质基准相似度评价结果r对照图谱s2~s16依次为:byt-2019091001d、byt-2019091002d、byt-2019091003d、byt-2019091604d、byt-2019091605d、byt-2019091606dbyt-2019091807d、byt-2019091808d、byt-2019091809d、byt-2019092310d、byt-2019092311d、byt-2019092312d、byt-2019092413d、byt-2019092414d、byt-20180515d以上15批次保元汤对应实物(冻干粉)指纹图谱生成对照指纹图谱r,以此比较15批次对应实物(冻干粉)相似度均大于0.85,不同批次间对应实物(冻干粉)的相似度良好。暂定供试品指纹图谱与对照指纹图谱的相似度不得低于0.85。1.3.3.2.5含量测定结合处方功效主治及处方药味在处方中的位置,综合确定含量测定指标。由于工艺研究确定水为溶剂,故保元汤物质基准中水溶性成分较多,其中君药人参主要成分为皂苷类成分,因此可测定其皂苷类成分人参皂苷rg1、rb1、re的总量的含量。臣药黄芪主要含有黄酮类、皂苷类等,虽为臣药,但其药理作用与保元汤物质基准的功效相关,故对黄芪中黄芪甲苷含量进行测定,以综合评价物质基准质量。①人参照高效液相色谱法《中国药典》2015年版四部(通则0512)测定a仪器与试药液相色谱仪:赛默飞-u3000;色谱柱:inertsustain-aq-c18(250mm×4.6,5μm);试剂:乙腈为色谱纯,水为超纯水;其他试剂均为分析纯。对照品:人参皂苷rg1、rb1、re的对照品(批号:110703-201832、批号:110704-201726、批号:110754-201827)均购自中国食品药品检定研究院)。b色谱条件的选择a)检测波长的选择精密称取人参皂苷rg1对照品、人参皂苷re对照品及人参皂苷rb1对照品,加甲醇配制成适宜的对照品溶液(人参皂苷rg1浓度为33.6024μg/ml,批号:110703-201731;人参皂苷re浓度为99.533μg/ml,批号:110704-201726;人参皂苷rb1浓度为58.506μg/ml,批号:1107544-201324),在190~500nm波长下进行扫描,可见人参皂苷rg1在202处有最大吸收峰、人参皂苷rb1在204处有最大吸收峰、人参皂苷re在203处有最大吸收峰。总结说明:人参皂苷rg1在202处有最大吸收峰、人参皂苷rb1在203处有最大吸收峰、人参皂苷re在204处有最大吸收峰,药典人参皂苷rg1、rb1、re的检测波长为203nm,人参皂苷rg1、rb1、re的的最大吸收峰包含药典人参皂苷rg1、rb1、re的检测波长,为兼顾人参皂苷rg1、rb1、re的测定及参考相关文献的色谱条件,故选用203nm作为优选检测波长。b)流动相的选择方法一:参照文献“四君子汤和理中丸方中人参皂苷的含量测定”色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为203nm,流速为1ml/min,流动相梯度洗脱见表46。表46流动相梯度洗脱表供试品溶液的制备取本品约1g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。结果:该方法的分离效果不好。方法二:参考文献“_双参片中人参皂苷rb1、rg1、re含量测定”色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为203nm,流速为1ml/min,具体梯度如下表47:表47流动相梯度洗脱表供试品溶液的制备取本品约1g,精密称定,加40氯仿回流1小时,放冷,残渣挥干,加30ml甲醇,加热回流2小时,放冷过滤,滤液蒸干,残渣加水溶解(分3次洗涤溶解,分别为15ml、10ml、10ml,合并洗液),加入3%氨水溶液1ml,用水饱和的正丁醇提取4次,每次35ml,合并正丁醇液,蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。实验结果:该方法的分离效果不好。方法三:参照《中国药典》2015年一部“人参”含量测定项下色谱条件,对方法一、二项下处理的供试品进样,色谱条件如下:色谱条件与系统用性试验以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相a,以水为流动相b,检测波长为203nm,具体梯度如下表48.表48流动相梯度洗脱表实验分析:在该梯度条件下,采用方法一供试品处理方法,色谱峰之间的分离度良好,因此供试品处理方法选择方法一,色谱条件选用《中国药典》人参项下含量测定色谱条件进行下一步考察。c供试品溶液制备方法考察a)提取方式的考察取本品(批号:byt-2019091001d)约1g(共3组),精密称定,加12.5ml水溶解,分别超声处理30min,回流处理30min,不做处理,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。精密吸取续滤液10μl,注入液相色谱仪,测定,计算,即得。结果见表49。表49提取方式考察的结果结果表明,不同的提取方式样品的含量相差不大,考虑提取方便,故选择直接处理。b)提取溶媒的考察取本品(批号:byt-2019091001d)约1g(共3组),精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液分别用石油醚、乙酸乙酯、水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,石油醚层、乙酸乙酯层、正丁醇层液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。精密吸取续滤液10μl,注入液相色谱仪,测定,计算,即得。结果如下表50。表50提取溶媒考察的结果结果表明:用水饱和的正丁醇液提取,人参皂苷rg1、rb1、re的总量总含量较高,故选择水饱和正丁醇为提取溶媒。c含量测定方法的初步确定色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为203nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000。按下表51中的规定进行梯度洗脱。表51梯度洗脱表对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得供试品溶液的制备取本品约1g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得。d方法学考察a)专属性试验人参皂苷rg1、rb1、re的总含量作为保元汤指标性成分,为考察药材之间是否相互干扰,按处方比列取药材同法制成阴性样品(批号:byt-rs-20190917-d01),按供试品的处理方法制备缺人参的阴性样品溶液进样记录色谱图。结果表明,阴性色谱中在与人参皂苷rg1、rb1、re相应的保留时间没有色谱峰,表明两味药之间测定无干扰,以本法测定本品中人参皂苷rg1、rb1、re的含量具有专属性。结果表明,阴性色谱中在与人参皂苷rg1、rb1、re相应的保留时间没有色谱峰,表明两味药之间测定无干扰,以本法测定本品中人参皂苷rg1、rb1、re的总含量具有专属性。b)线性关系考察精密吸取对照品混合溶液(人参皂苷rg1浓度为0.2016mg/ml,批号110703-201731;人参皂苷rg1浓度为0.223mg/ml,批号110704-201726;人参皂苷rg1浓度为0.2106mg/ml,批号110754-201324)2μl、4μl、8μl、12μl、16μl、20μl进样,记录色谱图,测得峰面积,以峰面积(a)对进样量(c)进行回归,得标准曲线。结果如下表52。表52人参皂苷rg1、rb1、re的总量线性关系人参皂苷rg1的回归方程:y=0.0041x+0.0447,人参皂苷rg1的总量相关系数:r=0.9997,人参皂苷rg1在403.2μg~4032μg范围内线性良好。人参皂苷re的回归方程:y=0.0017x+0.1657,人参皂苷re的相关系数:r=0.9994,人参皂苷re在0.446μg~4460μg范围内线性良好。人参皂苷rb1的回归方程:y=0.0031x+0.0597,人参皂苷rb1的相关系数:r=0.9999,人参皂苷rb1在421.2μg~4212μg范围内线性良好。c)仪器精密度取人参皂苷rg1、rb1、re的混合对照品溶液,按正文测定法项下操作,连续进样6次,测定结果见表53。表53仪器精密度试验结果(n=6)d)稳定性试验取本品(批号:byt-2019091807d)约1g,精密称定,按确定供试品溶液的制备和测定法项下操作,分别在制备后放置0、2、4、8、12、24小时进样测定,测定结果见下表54。表54稳定性试验结果e)重复性试验取本品(批号byt-2019091807d)约1g(共6份),精密称定,按正文供试品溶液制备和测定法项下进行操作。结果见下表55。表55重复性试验结果(n=6)f)加样回收试验取本品(批号:byt-2019091807d),精密称取约0.5g,共6份,分别精密加入人参皂苷rg1、rb1、re浓度分别为0.2016(批号:110703-201731)、0.223(批号:110704-201726)、0.2106(批号:110754-201324)mg/ml的混合对照品溶液1.1ml,按供试品溶液的制备方法制备样品溶液,依法测定,结果见下表56。表56加样回收试验结果(n=6)f含量测定方法确认以上的研究结果试验表明,保元汤对应实物(冻干粉)中人参皂苷rg1、rb1、re的总含量测定方法的专属性强,精密度、稳定性、重复性、加样回收等均符合规定,故此液相色谱条件可以用于保元汤对应实物(冻干粉)中的人参皂苷rg1、rb1、re的总含量测定,具体测定条件如下:色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为203nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000。按下表57中的规定进行梯度洗脱。表57梯度洗脱表对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得供试品溶液的制备取本品约1g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得。g多批次对应实物(冻干粉)含量测定按正文供试品溶液的制备和测定法项下操作,测定15批次对应实物(冻干粉)中人参皂苷rg1、rb1、re的总含量,结果见表58。表58不同批次对应实物(冻干粉)中人参皂苷rg1、rb1、re的总含量测定结果由上表可知:人参皂苷rg1、rb1、re的总含量的最小值为0.929mg/g,最大值为1.973mg/g,平均值为1.480mg/g,sd值为0.281,均值减3倍sd值为0.638mg/g,均值加3倍sd值为2.322mg/g。结合生产实际需要,故暂定人参皂苷rg1、rb1、re的总含量的范围为0.6mg/g~2.4mg/g。②黄芪液相色谱仪:赛默飞ultimate3000;色谱柱:glscienceslncaq-c18(5μm,250×4.6mm);welchaq-c18(5μm,250×4.6mm)等。上海越平科学仪器有限公司fa1104(万分之一)日本岛津科技有限公司auw1220d(十万分之一)乙腈为色谱纯:tedia生产;甲醇为色谱纯:tedia生产;磷酸为色谱纯,aladdin生产;甲醇为分析纯:常州市鸿盛精细化工有限公司;水为超纯水,成都浩康科技有限公司。黄芪甲苷对照品(批号:110781-201616,纯度:97.4%),购自中国食品药品检定研究院。含量测定方法的初步确定参照《中国药典》“黄芪颗粒”含量测定取本品1.0g,精密称定,精密加水50ml,称定重量,超声处理(功率720w,频率40khz)45分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取4次,每次25ml,合并正丁醇提取液,用氨试液洗涤3次,每次30ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。以十八烷基硅烷键合硅胶为填充剂;以乙腈-水(35:65)为流动相;蒸发光散射检测器检测。理论板数按黄芪甲苷峰计算应不低于4000。色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈-水(35:65)为流动相;蒸发光散射检测器检测。理论板数按黄芪甲苷峰计算应不低于4000。对照品溶液的制备取黄芪甲苷对照品适量,精密称定,加甲醇制成每lml含0.4mg的溶液,即得。供试品溶液的制备取本品1.0g,精密称定,精密加水50ml,称定重量,超声处理(功率720w,频率40khz)30分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取4次,每次25ml,合并正丁醇提取液,用氨试液洗涤3次,每次30ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。测定法精密吸取对照品溶液10μl、20μl,供试品溶液20μl,注人液相色谱仪,测定,以外标两点法对数方程计算,即得。a方法学考a)专属性试验黄芪甲苷含量作为保元汤指标性成分之一,为考察药材之间是否相互干扰,按处方比列取药材同法制成阴性样品(批号:byt-hq-20190917-d01),按供试品的处理方法制备缺黄芪的阴性样品溶液进样记录色谱图。结果表明,阴性色谱中在与黄芪甲苷相应的保留时间没有色谱峰,表明味药之间测定无干扰,以本法测定本品中黄芪甲苷含量具有专属性。a)线性关系考察精密吸取浓度为0.5018mg/ml黄芪甲苷对照品(批号:110781-201616)溶液2μl、4μl、8μl、12μl、16μl、20μl,注入液相色谱仪,照高效液相色谱法(《中国药典》2015年版四部通则0512)测定。以进样量(μg)为横坐标,峰面积为纵坐标,绘制标准曲线。结果如下表59。回归方程:y=4.7063x-4.5398相关系数:r=r=0.9972结果表明黄芪甲苷在1.0036~10.036μg范围内线性良好表59黄芪甲苷线性关系b)仪器精密度取黄芪甲苷对照品溶液,按正文测定法项下操作,连续进样6次,测定结果见表60。表60仪器精密度试验结果(n=6)结果表明:rsd<2%,仪器精密度良好。c)稳定性试验取本品(批号:byt-2019091807d)1g密称定,按确定供试品溶液的制备和测定法项下操作,分别在制备后放置0、2、4、8、12、24小时进样测定,测定结果见下表61。表61稳定性试验结果结果表明:rsd<2%,样品稳定性度良好。d)重复性试验实验人员(a)操作,取本品(批号:byt-2019091807d)约1g(共5份),精密称定,按正文供试品溶液制备和测定法项下进行操作。结果见下表62。表62重复性试验结果(n=6)结果表明,rsd<3.0%,说明本方法的重复性良好。e)加样回收试验精密称取取物质基准样品(黄芪甲苷含量0.661mg/g)0.5g,共6份,精密加入黄芪甲苷对照品溶液(浓度:0.5018mg/ml,批号:110781-201616)0.28ml,按供试品溶液的制备方法制备样品溶液,依法测定,结果见下表63。表63黄芪甲苷准确度试验结果(n=6)结果,平均回收率为98.32%,rsd<2.26%,表明本方法准确度良好。f含量测定方法确认以上的研究结果试验表明,保元汤对应实物(冻干粉)中黄芪甲苷的含量测定方法的专属性强,稳定性、重复性、准确度等均符合规定,且色谱条件的耐用性良好,故此液相色谱条件可以用于保元汤对应实物(冻干粉)中的黄芪甲苷的含量测定,具体测定条件如下:色谱条件与系统适用性试验十八烷基硅烷键合硅胶为填充剂;以乙腈-水(35:65)为流动相;蒸发光散射检测器检测。理论板数按黄芪甲苷峰计算应不低于4000。对照品溶液的制备取黄芪甲苷对照品适量,精密称定,加甲醇制成每lml含0.5mg的溶液,即得。供试品溶液的制备取本品1.0g,精密称定,精密加水50ml,称定重量,超声处理(功率720w,频率40khz)45分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取4次,每次25ml,合并正丁醇提取液,用氨试液洗涤3次,每次30ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。测定法测定法精密吸取对照品溶液10μl、20μl,供试品溶液20μl,注人液相色谱仪,测定,以外标两点法对数方程计算,即得。g多批次对应实物(冻干粉)含量测定按正文供试品溶液的制备和测定法项下操作,测定15批次对应实物(冻干粉)中黄芪甲苷的含量,结果见表64。表6415批次对应实物(冻干粉)中黄芪甲苷的含量结果表明:15批对应实物(冻干粉)黄芪甲苷范围0.479~1.037mg/g,平均含量0.772mg/g,减3sd值为0.19mg/g,加3sd值为1.35mg/g,同时考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计算,每1g含黄芪甲苷(c41h68o14)应为0.10~1.40mg(±3倍sd值)。1.3.4对应实物的质量分析1.3.4.1对应实物制备将处方中药味,按照确定工艺,详见“工艺研究”,将处方中各15批合格饮片进行物质基准的制备,每批投料20倍日处方量,约30g,平行制备15批次物质基准,具体制备结果如下表65。表65十五批物质基准1.3.4.2对应实物测定方法(1)出膏率测定:测定方法详见“工艺研究”。(2)水分测定:参照《中国药典》2015年版四部通则0832水分测定第二法,对不同批次物质基准水分进行检测,取供试品2g,平铺于干燥至恒重的扁形称量瓶中,厚度不超过5mm,疏松供试品不超过10mm,精密称定,开启瓶盖在105℃干燥5小时,将瓶盖盖好,移置干燥器中,放冷30分钟,精密称定,再在上述温度干燥1小时,放冷,称重,至连续两次称重的差异不超过5mg为止。根据减失的重量,计算供试品中含水量小于12(%)。(3)浸出物测定:参照2015年版《中国药典》通则2201醇溶性热浸法测定:取本品适量,研细,取约2g,精密称定,置100ml的锥形瓶中,精密加乙醇50ml,密塞,称定重量,静置1小时后,采用热浸法进行浸出物测定,精密量取滤液25ml置已干燥至恒重的蒸发皿中,在水浴上蒸干后,于105℃干燥3小时,置干燥器中冷却30分钟,迅速精密称定重量。计算供试品中醇溶性浸出物的含量18.0%~81.0%(%)。(4)指纹图谱测定:药材、饮片、中间体、对应实物测定方法详见“8.4质量标准正文”,根据15批次物质基准指纹图谱测定结果,标出12个共有峰,并生成对照指纹图谱,计算相似度。药材、饮片、中间体供试品制备方法如下:a甘草药材溶液:按处方称取甘草药材粉末(过四号筛)2.5g,精密称定,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。b甘草饮片溶液:按处方称取甘草饮片粉末(过四号筛)2.5g,精密称定,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。c人参饮片溶液:按处方称取人参饮片粉末(过四号筛)2.5g,精密称定,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。d中间体(提取液):精密量取提取液5ml,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。e中间体(浓缩液):精密量取浓缩液2ml,置具塞锥形瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加水20ml溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml氨试液萃取1次,弃去氨试液,正丁醇层蒸干,残渣加2ml水溶解。经装有10mld101大孔吸附树脂的色谱柱,先用50ml水洗脱,后用95%乙醇100ml洗脱,收集95%乙醇洗脱液,蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,过滤,即得。(5)含量测定:对应实物人参皂苷rg1、rb1、re的总含量、黄芪甲苷测定方法详见“8.4质量标准正文”。中间体供试品制备方法如下:①提取液人参皂苷rg1、rb1、re的总含量:精密吸取提取液25ml,置分液漏斗中,加氯仿提取3次,每次15ml,弃氯仿层,水层用水饱和的正丁醇提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次每次30ml,弃氨试液层,正丁醇层用正丁醇饱和水20ml洗涤1次,弃水层,正丁醇液蒸干,残渣加适量甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度即得。黄芪甲苷含量:精密量取提取液25ml,超声处理30min,用水饱和正丁醇溶液提取4次,每次25ml,合并正丁醇液,用氨试液振摇提取3次每次30min,弃氨试液层,收集正丁醇液液,蒸干,残渣加甲醇使之溶解,转移至5ml的容量瓶中,加甲醇定容至刻度即得。②浓缩液人参皂苷rg1、rb1、re的总含量:精密吸取提取液10ml,置分液漏斗中,加氯仿提取3次,每次15ml,弃氯仿层,水层用水饱和的正丁醇提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次每次30ml,弃氨试液层,正丁醇层用正丁醇饱和水20ml洗涤1次,弃水层,正丁醇液蒸干,残渣加适量甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度即得。黄芪甲苷含量:精密量取提取液10ml于25ml的容量瓶中,加水定容至刻度,超声处理30min,用水饱和正丁醇溶液提取4次,每次25ml,合并正丁醇液,用氨试液振摇提取3次每次30min,弃氨试液层,收集正丁醇液液,蒸干,残渣加甲醇使之溶解,转移至5ml的容量瓶中,加甲醇定容至刻度即得。1.3.4.3测定结果结果汇总表如下表66。1.3.4.4对应实物关键质量属性范围的确定(1)出膏率15批次物质基准出膏率范围9.85~17.52%,平均出膏率13.97%,均在3sd浮动范围6.65~21.28%;均值±70%浮动范围9.78%~18.16%。暂定出膏率范围为6.00~22.00%。(2)水分15批次物质基准水分范围6.3%~11.8%,平均水分8.7%,均值±3sd值范围为4.05%~13.38%,故暂定水分不得过12.0%。(3)浸出物15批次物质基准浸出物范围27.8%~70.1%,平均浸出物49.47%,sd值为0.1,均值±3sd值范围为18.43%~80.52%暂定浸出物范围为18.0%~81.0%。(4)指纹图谱15批次保元汤物质基准指纹图谱生成对照指纹图谱,依次比较15批次物质基准相似度均大于0.85,不同批次间物质基准的相似度良好,暂定指纹图谱相似度限度为≥0.85。(5)含量测定15批物质基准人参皂苷rg1、rb1、re的总含量范围0.929mg/g~1.973mg/g,平均含量1.480mg/g,均在±30%及3sd范围内;参照±30%及3sd浮动范围,同时考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计算,每1g含人参以人参皂苷rg1(c42h72o14)、rb1(c54h92o23)、re(c48h82o18)的总含量计,应为0.6mg/g~2.4mg/g。15批物质基准黄芪甲苷范围0.479mg/g~1.037mg/g,平均含量0.772mg/g,均在3sd范围内,参照±30%及3sd浮动范围,同时考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计算,每1g含黄芪以黄芪甲苷(c41h68o14)计,应为0.10mg/g~1.40mg/g。1.4保元组合物检测方法起草说明1.4.1处方本方出自于《简明医彀》(明·孙志宏)。详细研究见“资料5处方考证及历史沿革资料”。1.4.2制法根据经典名方目录中记载的方法进行煎煮。采用电子煎药罐(容量9l)煎煮,以出膏率、人参皂苷rg1、rb1、re的总含量、黄芪甲苷提取含量、指纹图谱为指标,研究物质基准饮片前处理、加热方式、煎煮时间、滤过及干燥工艺等进行研究相关参数,从而确定保元汤物质基准工艺。按照确定工艺,将处方量放大20倍进行投料,提取15批煎液,经滤过、浓缩、干燥。得到15批物质基准对应实物,结果显示物质基准制备工艺稳定、可行。保元汤物质基准制法为:人参3.69g、黄芪7.38g、甘草1.85g、肉桂0.74g、生姜一片,以上四味药,加生姜,将处方量放大20倍,即人参74g,黄芪148g,甘草37g,肉桂25g,生姜20片,按中药配方颗粒根茎类药材加水量标准,加水量为处方量的7倍,即1920ml,浸泡40min,煎煮1h,200目滤布过滤,滤液低温真空浓缩至与投料量比为1﹕3的浸膏,低温冷冻干燥,即得物质基准对应实物,置低温干燥处储存。(具体研究见“工艺研究”)。1.4.3性状根据多批次样品的实际观察情况,暂定本品为棕色至棕褐色粉末,气微香,味微苦甜。1.4.4鉴别本品由四味中药制成,系用水煎煮工艺制成的复方制剂,选用了专属性强,快速、简便、重现性好的薄层鉴别方法,对方中人参、黄芪、甘草、肉桂的主要特征成分进行鉴别试验研究,确定人参、黄芪、甘草、肉桂纳入质量标准。(1)人参人参主要成分为皂苷类、糖类、挥发性成分、有机酸及其酯、甾醇及其苷、黄酮类、木质素、无机元素及维生素类等。确定薄层鉴别方法:取本品粉末1g,加三氯甲烷40ml,加热回流1小时,弃去三氯甲烷液,药渣挥干溶剂,加水0.5ml搅拌湿润,加水饱和正丁醇10ml,超声处理30分钟,吸取上清液加3倍量氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取人参对照药材1g,同法制成对照药材溶液。再取人参皂苷rb1对照品、人参皂苷re对照品、人参皂苷rf对照品及人参皂苷rg1对照品,加甲醇制成每1ml各含2mg的混合溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各1~2μ1,分别点于同一硅胶g薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15:40:22:10)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,分别置日光和紫外光灯(365nm)下检视,供试品色谱中,在与对照品色谱相应的位置上,显相同的蓝紫色斑点,阴性色谱在对照色谱相应的位置上无斑点。通过对低温、常温、高湿环境及不同厂家的硅胶g薄层板进行耐用性考察,结果不同影响因素下,此人参薄层鉴别条件耐用性良好。不同批次物质基准的供试品色谱中,在与对照药材色谱和对照品色谱相应位置上,显相同颜色斑点。故将此薄层方法列入正文。(2)黄芪黄芪主要化学成分为皂苷类和黄酮类,除此之外还含有多糖等。根据以上化学成分对黄芪进行薄层色谱的摸索实验,确定方法。确定薄层鉴别方法:取本品1.5g,加水25ml,超声处理30分钟,滤过,滤液用水饱和正丁醇振摇提取2次,每次25ml,合并正丁醇提取液,用氨试液洗涤2次,每次30ml,分取正丁醇液蒸干,残渣加甲醇lml使溶解,作为供试品溶液。另取黄芪对照药材1.5g,加水50ml,煎煮30分钟,滤过,滤液同法制成对照药材溶液。再取黄芪甲苷对照品,加甲醇制成每lml含2mg的溶液,作为对照品溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015年版四部通则0502)试验,吸取对照品溶液、对照药材溶液、供试品溶液各10μl,分别点于同一硅胶g薄层板上,以三氯甲烷_甲醇-水(13:7:2)10℃以下放置的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点;置紫外光灯(365nm)下显检视,显相同颜色的荧光斑点。通过对低温、常温、高湿环境及不同厂家的硅胶g薄层板进行耐用性考察,结果不同影响因素下,此人参薄层鉴别条件耐用性良好。不同批次物质基准的供试品色谱中,在与对照药材色谱和对照品色谱相应位置上,显相同颜色斑点。故将此薄层方法列入正文。(3)甘草甘草主要化学成分为三萜类和黄酮类,除此之外还含有多糖、有机酸和香豆素等。根据以上化学成分对甘草进行薄层色谱的摸索实验,确定方法。确定薄层鉴别方法:取对应实物(冻干粉)及缺甘草阴性冻干粉1g,加水40ml,超声30分钟,滤过,用乙醚振摇提取2次,每次10ml,弃去乙醚液,水液用水饱和正丁醇振摇提取3次,每次20ml,合并正丁醇液,用氨试液振摇提取2次,每次30ml,合并氨试液,用盐酸调ph值至3~4。再用乙酸乙酯振摇提取2次,每次30ml,合并乙酸乙酯液,蒸干,残渣加甲醇1ml使溶解,作为供试品溶液。另取炙甘草饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。取甘草苷对照品适量,加70%乙醇制成每1ml含0.5mg的溶液,作为对照品溶液。照薄层色谱法2015版四部(通则0502)试验,吸取上述三种溶液各10μl,分别点于同一用1%氢氧化钠溶液制备的硅胶g薄层板上,以三氯甲烷-甲醇-水(13:7:2)5~10℃放置的下层液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色淸晰,置日光及紫外光灯(365nm)下检视。通过对低温、常温、高湿环境及不同厂家的硅胶g薄层板进行耐用性考察,结果不同影响因素下,此炙甘草薄层鉴别条件耐用性良好。不同批次物质基准的供试品色谱中,在与对照药材色谱和对照品色谱相应位置上,显相同颜色斑点。故将此薄层方法列入正文。(4)肉桂肉桂主要含有桂皮醛、肉桂酸、二萜及其糖苷、黄烷醇及其多聚体,除此之外还含有黄酮类、多酚类等。根据以上化学成分对肉桂进行如下薄层色谱的摸索实验。确定薄层鉴别方法:取对应实物(冻干粉)及缺肉桂阴性冻干粉各1g,加水10ml使其溶解,用乙醚振摇提取1次,每次25ml,合并乙醚液,挥干,残渣加乙醚2ml使溶解,作为供试品溶液及缺肉桂阴性样品溶液。另取肉桂饮片1g,加水50ml,煎煮30分钟,滤过,同法制成对照药材溶液。再取肉桂酸对照品,用50%甲醇制成1ml含0.2mg的溶液,作为对照品溶液。照薄层色谱法(《中国药典》2015版四部通则0502)试验,吸取上述四种溶液各3μl,点于同一硅胶gf254薄层板上,以环己烷-乙醚-冰醋酸(5:5:0.3)为展开剂,展开,取出,晾干,置紫外光灯(254nm)下检视,在供试品色谱中,在与对照药材色谱和对照品色谱相应的位置上,显相同颜色的斑点或荧光斑点。故将此薄层方法列入正文。1.4.5检查(1)水分照水分测定法(《中国药典》2015年版四部通则0832第二法),测定15批物质基准水分最小值为6.3%,最大值为11.8%,平均值为8.7%,平均值的±3sd值为4.05%~13.38%,故暂定干膏水分不得过12.0%。1.4.6浸出物中药材中的化学成分或大类成分多数可以在水或醇中溶出,进而进行浸出物测定。由于保元汤物质基准是水提取,选择水为溶剂建立浸出物测定意义不大,难以反映内在质量,故选溶剂时,要考虑大类成分的溶解度,选择乙醇进行浸出物测定,以控制物质基准的质量。照浸出物测定法(《中国药典》2015年版四部通则2201醇溶性热浸法)测定15批物质基准浸出物最小值为27.79%,最大值为70.07%,平均值为49.47%,平均值的±3sd值为18.43%~80.52%。考虑到实际生产过程中的各种影响因素等,暂定本品醇溶性浸出物的含量为18.0%~81.0%。1.4.7指纹图谱由于单纯的指标成分的定性鉴别和定量分析,难以反映物质基准质量的优劣。作为衡量物质基准对应实物是否与物质基准基本一致的标准参照物,其质量应加强专属性鉴别和多成分、整体质量控制。因而物质基准质量控制要重视以指纹图谱为主的整体质量控制。对方法流动相组成及梯度、供试品制备方法进行摸索,并对物质基准指纹图谱中各色谱峰进行药味归属、峰指认及参照物的选择(选择保留时间适中,响应值较高,达到基线分离的毛蕊异黄酮葡萄糖苷作为参照物),初步确定指纹图谱测定方法。对初步确定的指纹图谱方法进行方法学验证,方法的专属性良好,空白溶剂无干扰,同时基本满足了信息量最大的原则,且方法精密度、重复性、稳定性(24小时内)均符合规定(各色谱峰相对保留时间rsd小于2%),色谱图中各共有峰峰型尖锐,对称,分离度良好,故将此方法确定为指纹图谱测定方法列入标准正文:色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂;色谱柱:inertsustain-aq-c18,以乙腈为流动相a,以0.1%磷酸溶液为流动相b,流速为1ml/min,检测波长为210nm,具体梯度见下表67。表67流动相梯度洗脱表参照物溶液的制备取毛蕊异黄酮葡萄糖苷对照品适量,精密称定,加50%甲醇制成每1ml含毛蕊异黄酮葡萄糖苷0.2068mg/ml的溶液,即得。供试品溶液的制备取本品约2.5g,精密称定,置50ml三角锥瓶中,加50%甲醇50ml超声45min,滤过,滤液减压回收溶剂至近干,残渣加20ml水溶解,用水饱和的正丁醇溶液萃取2次,每次25ml,合并正丁醇溶液,用30ml的氨试液萃取1次,弃去氨试液,正丁醇层溶液挥干,残渣用2ml水溶解,经装有10mld101大孔吸附树脂的柱色谱,先用50ml水洗脱,后用95%乙醇100ml洗脱,将95%乙醇洗脱液蒸干,残渣加50%甲醇溶解并转移至10ml容量瓶中,定容至刻度,摇匀,滤过,取续滤液,即得。测定法分别精密吸取参照物溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录70min的色谱图,即得。按照上述确定方法对15批物质基准进行测定,并对所得指纹图谱进行分析,选择15批物质基准指纹图谱中稳定性较好、响应值适宜的色谱峰为共有峰,共标定了12个共有峰。采用国家药典委员会颁布的《中药色谱指纹图谱相似度评价软件系统2012年版》,以15批保元汤物质基准对应实物(干膏粉)hplc图谱色谱峰进行自动匹配,形成共有模式图,并建立对照图谱。以共有峰对15批样品与对照指纹图谱的相似度进行计算,均大于0.85,故选择指纹图谱作为保元汤物质基准的评价标准,暂定本品与保元汤物质基准对照指纹图谱比较,其相似度不得低于0.85。1.4.8含量测定结合处方功效主治及处方药味在处方中的位置,综合确定含量测定指标。由于工艺研究确定水为溶剂,故保元汤物质基准中水溶性成分较多,其中君药人参主要成分为单萜及其苷类成分,因此可测定其苷类成分人参皂苷rg1、rb1、re的总量的含量。臣药黄芪主要含有黄酮类、皂苷类等,虽为臣药,但其药理作用与保元汤物质基准的功效相关,故对黄芪中黄芪甲苷含量进行测定,以综合评价物质基准质量。人参对人参皂苷rg1、rb1、re的总含量测定方法流动相组成及比例、供试品制备方法、检测波长进行摸索,初步确定含量测定方法。对初步确定的方法进行方法学验证,方法的专属性良好,缺人参阴性样品无干扰,目标峰纯度合格,人参皂苷rg1在进样量为403.2μg~4032μg的范围内线性良好。人参皂苷re在进样量为466μg~4660μg的范围内线性良好。,人参皂苷rb1在进样量为421.2μg~4212μg的范围内线性良好。且方法精密度(rsd<2.0%)、重复性(rsd<3.0%)、稳定性(24小时内rsd<3.0%)、准确度(平均回收率为96.656%,rsd为1.98%)均符合规定,黄芪甲苷铵在进样量为1.0036μg~10.036μg的范围内线性良好。且方法精密度(rsd<2.0%)、重复性(rsd<3.0%)、稳定性(24小时内rsd<3.0%)准确度(平均回收率为98.32%,rsd为2.26%)均符合规定。色谱条件的耐用性良好。故此液相色谱条件可以用于保元汤对应实物(冻干粉)中的人参皂苷rg1、rb1、re的总含量的含量、黄芪甲苷含量测定,具体测定条件如下:人参色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为203nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000。按下表68中的规定进行梯度洗脱。表68梯度洗脱表对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得供试品溶液的制备取本品约1g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得。本品按干燥品计算,每1g含人参以人参皂苷rg1(c42h72o14)、rb1(c54h92o23)、re(c48h82o18)的总含量计,应为0.6mg/g~2.4mg/g。色谱条件与系统适用性试验以十八烷基硅烷键合硅胶为填充剂,以乙腈为流动相a,以水溶液为流动相b,检测波长为203nm,理论板数按人参皂苷rg1的总量峰计算应不低于5000。按下表69中的规定进行梯度洗脱。表69梯度洗脱表对照品溶液的制备取人参皂苷rg1、rb1、re对照品适量,精密称定,加甲醇制成每1ml含人参皂苷rg1、rb1、re的0.2mg的混合溶液,即得供试品溶液的制备取本品约1g,精密称定,加12.5ml水溶解,置分液漏斗中,加氯仿提取3次,每次15ml,弃去氯仿层液,溶液用水饱和的正丁醇溶液提取3次,每次30ml,合并正丁醇液,用氨试液洗涤2次,每次30ml,弃氨试液层,正丁醇液用水饱和的正丁醇溶液水层溶液20ml,洗涤1次,弃水层溶液,正丁醇液蒸干,残渣加甲醇溶解,转移至5ml的容量瓶中,加甲醇定容至刻度,摇匀,即得。测定法分别精密吸取对照品溶液和供试品溶液各10μl,注入液相色谱仪,测定,记录110min的色谱图,即得。本品按干燥品计算,每1g含人参以人参皂苷rg1(c42h72o14)、rb1(c54h92o23)、re(c48h82o18)的总含量计,应为0.6mg/g~2.4mg/g。15批物质基准人参皂苷rg1、rb1、re的总含量范围0.929mg/g~1.973mg/g,平均含量1.480mg/g,均在±30%及3sd范围内;参照±30%及3sd浮动范围,同时考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计算,每1g含人参以人参皂苷rg1(c42h72o14)、rb1(c54h92o23)、re(c48h82o18)的总含量计,应为0.6mg/g~2.4mg/g。黄芪色谱条件与系统适用性试验十八烷基硅烷键合硅胶为填充剂;以乙腈-水(35:65)为流动相;蒸发光散射检测器检测。理论板数按黄芪甲苷峰计算应不低于4000。对照品溶液的制备取黄芪甲苷对照品适量,精密称定,加甲醇制成每lml含0.5mg的溶液,即得。供试品溶液的制备取本品1.0g,精密称定,精密加水50ml,称定重量,超声处理(功率720w,频率40khz)45分钟,再称定重量,用水补足减失的重量,摇匀,滤过,精密量取续滤液25ml,用水饱和的正丁醇振摇提取4次,每次25ml,合并正丁醇提取液,用氨试液洗涤3次,每次30ml,弃去氨试液洗液,分取正丁醇液蒸干,残渣用甲醇溶解并转移至5ml量瓶中,加甲醇稀释至刻度,摇匀,即得。本品按干燥品计算,每1g含黄芪以黄芪甲苷(c41h68o14)计,应为0.10mg/g~1.40mg/g。15批物质基准黄芪甲苷范围0.479mg/g~1.037mg/g,平均含量0.772mg/g,均在3sd范围内,参照±30%及3sd浮动范围,同时考虑到实际生产过程中的各种影响因素等,暂定本品按干燥品计算,每1g含黄芪以黄芪甲苷(c41h68o14)计,应为0.10mg/g~1.40mg/g。虽然,上文中已经用一般性说明、具体实施方式及试验,对本发明作了详尽的描述,但在本发明基础上,可以对之作出一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1