CD28T细胞培养物、组合物及其使用方法与流程
cd28 t细胞培养物、组合物及其使用方法
背景
1.技术领域
2.本公开提出了改善t细胞疗效的方法。一方面,本公开进一步提出了增强和预测t细胞产物的最终倍数扩增、cd8:cd4 t细胞的比率、相对最终端粒长度和克隆丰富度的方法。本公开还提出了有此需要受试者的癌症治疗方法以及透过本文描述的方法产生的t细胞群。
3.2.背景
4.免疫疗法已成为治疗癌症的非常有前途的方法。免疫疗法可细分为细胞疗法和小分子/抗体疗法。在细胞疗法领域内,嵌合抗原受体t(car
‑
t)细胞疗法已对液态肿瘤显示出强大的临床疗效,而基于t细胞受体t(tcr
‑
t)细胞的疗法已对各种实体瘤适应症显示出具有前景的早期效果。临床产品的疗效可能受到其体内特性驱动,这些体内特性大多可能在离体制造过程中印记。
5.us 8,383,099描述了一种透过培养自体t细胞以及使用okt3抗体、il
‑
2和饲养淋巴细胞来扩增培养t细胞从而促进受试者癌症消退的方法。
6.us 9,074,185描述了一种产生t细胞输注产物来促进受试者癌症消退的方法,该方法包括培养自体t细胞、使培养t细胞富集cd8+ t细胞、使用okt3抗体、il
‑
2和饲养淋巴细胞来扩增培养t细胞数量从而提供扩增数量的t细胞。
7.对于癌症患者,仍然需要改善t细胞的疗效和act的结果。权利要求中所表征的实施方案提供了该技术问题的解决方案。简要概述
8.本公开提出了产生疗效改善的t细胞的方法,例如:
·
从患者或供体中获得t细胞群,
·
确定所获得t细胞群中cd28+ cd8+ t细胞的百分比,
·
用抗cd3抗体和/或抗cd28抗体激活确定的t细胞群,以及
·
其中,确定的细胞群包括至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%或至少约99%的cd28+ cd8+ t细胞。
9.本公开进一步提出了产生疗效改善的t细胞的方法,例如:
·
从患者或供体中获得t细胞群,
·
确定所获得t细胞群中cd28+ cd8+ t细胞的百分比,
·
在不存在抗cd28抗体的情况下,用抗cd3抗体激活确定的t细胞群,以及
·
其中确定的细胞群包括小于约50%、小于约45%、小于约40%、小于约35%、小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%的cd28+ cd8
+ t细胞。
10.本公开进一步提出了产生疗效改善的t细胞的离体方法,例如:
·
确定所分离t细胞群中cd28+ cd8+ t细胞的百分比,
·
用抗cd3抗体和/或抗cd28抗体激活确定的t细胞群,以及
·
前提是,确定的细胞群包括至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%或至少99%的cd28+ cd8+ t细胞。
11.本公开进一步提出了产生疗效改善的t细胞的离体方法,例如:
·
确定所分离t细胞群中cd28+ cd8+ t细胞的百分比,
·
在不存在抗cd28抗体的情况下,用抗cd3抗体激活确定的t细胞群,以及
·
前提是,確定的細胞群包括小於50%、小於45%、小於40%、小於35%、小於30%、小於25%、小於20%、小於15%、小於10%、小於9%、小於8%、小於7%、小於6%、小於5%、小於4%、小於3%、小於2%或小於1%的cd28+cd8+ t細胞。
12.一方面,用病毒载体转导激活的t细胞群,并扩增转导的t细胞群。另一方面,转导和扩增可在存在至少一种细胞因数的情况下进行。
13.另一方面,本公开涉及用于产生改善免疫疗法疗效的t细胞的方法,包括:
·
从患者或供体中获得cd8+ t细胞群,
·
确定所获得细胞群中cd28+ cd8+ t细胞的百分比,
·
用抗cd3抗体和抗cd28抗体激活确定的细胞群,以及
·
其中,确定的细胞群包括至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%或至少约99%的cd28+ cd8+ t细胞,
·
用病毒载体转导激活的t细胞群,和
·
扩增转导的t细胞群。
14.另一方面,本公开涉及用于产生改善免疫疗法疗效的t细胞的离体方法,包括:
·
确定所分离cd8+ t细胞群中cd28+ cd8+ t细胞的百分比,
·
用抗cd3抗体和抗cd28抗体激活确定的细胞群,以及
·
前提是,确定的细胞群包括至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少91%、至少92%、至少93%、至少94%、至少95%、至少96%、至少97%、至少98%或至少99%的cd28+ cd8+ t细胞,
·
用病毒载体转导激活的t细胞群,和
·
扩增转导的t细胞群。
15.另一方面,本公开涉及用于产生改善免疫疗法疗效的t细胞的方法,包括:
·
从患者或供体中获得cd8+ t细胞群,
·
确定所获得细胞群中cd28+ cd8+ t细胞的百分比,
·
在不存在抗cd28抗体的情况下,用抗cd3抗体激活确定的细胞群,但是前提是,确定的细胞群包括小于约50%、小于约45%、小于约40%、小于约35%、小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约9%、小于约8%、小于约7%、小于约
6%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%的cd28+ cd8+ t细胞,
·
用病毒载体转导激活的t细胞群,和
·
扩增转导的t细胞群。
16.另一方面,本公开涉及用于产生改善免疫疗法疗效的t细胞的离体方法,包括:
·
确定所分离cd8+ t细胞群中cd28+ cd8+ t细胞的百分比,
·
在不存在抗cd28抗体的情况下,用抗cd3抗体激活确定的细胞群,但是前提是,确定的细胞群包括小于约50%、小于约45%、小于约40%、小于约35%、小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%的cd28+ cd8+ t细胞,
·
用病毒载体转导激活的t细胞群,和
·
扩增转导的t细胞群。
17.另一方面,转导和扩增可在存在至少一种细胞因数的情况下进行。
18.一方面,激活可能包括用抗cd3抗体和抗cd28抗体将t细胞固定于固相支撑物上。
19.另一方面,抗cd3抗体和/或抗cd28抗体各自的浓度为约0.1μg/ml至约10.0μg/ml、约0.1μg/ml至约8.0μg/ml、约0.1μg/ml至约6.0μg/ml、约0.1μg/ml至约4.0μg/ml、约0.1μg/ml至约2.0μg/ml、约0.1μg/ml至约1.0μg/ml、约0.1μg/ml至约0.5μg/ml、约0.5μg/ml至约10.0μg/ml、约2μg/ml至约8μg/ml、约3μg/ml至约7μg/ml、约2μg/ml至约5μg/ml、约0.5μg/ml至约2.0μg/ml或约0.5μg/ml至约2.5μg/ml。
20.另一方面,激活可能在约1小时至约120小时、约1小时至约108小时、约1小时至约96小时、约1小时至约84小时、约1小时至约72小时、约1小时至约60小时、约1小时至约48小时、约1小时至约36小时、约1小时至约24小时、约2小时至约24小时、约4小时至约24小时、约6小时至约24小时、约8小时至约24小时、约10小时至约24小时、约12小时至约24小时、约12小时至约72小时、约24小时至约72小时、约6小时至约48小时、约24小时至约48小时、约6小时至约72小时或约1小时至约12小时的时间段内进行。
21.另一方面,至少一种细胞因数可选自白介素(il)
‑
2、il
‑
7、il
‑
10、il
‑
12、il
‑
15、il
‑
21或其组合物。
22.另一方面,至少一种细胞因数包括il
‑
7、il
‑
15或il
‑
7和il
‑
15的组合物。
23.另一方面,il
‑
7的浓度为约1ng/ml至90ng/ml、约1ng/ml至80ng/ml、约1ng/ml至70ng/ml、约1ng/ml至60ng/ml、约1ng/ml至50ng/ml、约1ng/ml至40ng/ml、约1ng/ml至30ng/ml、约1ng/ml至20ng/ml、约1ng/ml至15ng/ml、约1ng/ml至10ng/ml、约2ng/ml至10ng/ml、约4ng/ml至10ng/ml、约6ng/ml至10ng/ml或约5ng/ml至10ng/ml。
24.另一方面,il
‑
15的浓度可能为约5ng/ml至500ng/ml、约10ng/ml至400ng/ml、约15ng/ml至300ng/ml、约5ng/ml至200ng/ml、约5ng/ml至150ng/ml、约5ng/ml至100ng/ml、约10ng/ml至100ng/ml、约20ng/ml至100ng/ml、约30ng/ml至100ng/ml、约40ng/ml至100ng/ml、约50ng/ml至100ng/ml、约60ng/ml至100ng/ml、约70ng/ml至100ng/ml、约80ng/ml至100ng/ml、约90ng/ml至100ng/ml、约10ng/ml至50ng/ml、约1ng/ml至50ng/ml、约5ng/ml至50ng/ml或约20ng/ml至50ng/ml。
25.另一方面,转导可能在约1小时至120小时、约12小时至96小时、约24小时至96小时、约24小时至72小时、约10小时至48小时、约1小时至36小时、约1小时至24小时、约2小时
至24小时、约4小时至24小时、约6小时至24小时、约8小时至24小时、约10小时至24小时、约1小时至12小时、约14小时至24小时、约1小时至12小时、约6至约18小时的时间段内进行。
26.另一方面,病毒载体可能是表达t细胞受体(tcr)的逆转录病毒载体。
27.另一方面,病毒载体可能是表达tcr的慢病毒载体。
28.另一方面、扩增可能在约1天至约30天、约5至约30天、约1天至约25天、约2天至约20天、约5天至约15天、约2天至约10天、约3天至约15天、约3天至约20天、约4天至约10天、约5天至约10天、约6天至约10天、约7天至约25天、约8天至约25天或约9天至约12天的时间段内进行。
29.另一方面,本公开涉及一种用于产生改善过继免疫疗法疗效的t细胞的方法,包括:例如,从患者或供体中获得cd8+ t细胞群、从所得细胞群中分离cd28+ cd8+ t细胞(其中分离的细胞包含至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%或至少约99%的cd28+ cd8+ t细胞)、用抗cd3抗体和抗cd28抗体激活分离的细胞、用病毒载体转导激活的细胞群以及扩增转导的细胞群(其中转导和扩增可在存在至少一种细胞因数的情况下进行)。
30.另一方面,本公开涉及透过本公开的方法产生的t细胞。
31.另一方面,本公开涉及可用本公开的方法获得的t细胞,优选为t细胞群,更优选为基因转导的t细胞。本公开的另一方面,t细胞,优选为t细胞群,更优选为基因转导的t细胞可使用本公开的方法直接获得。
32.一方面,透过本文所述方法提出的包含至少约50%cd28+ cd8+ t细胞的基因转导t细胞与从包含少于约50%的cd28+ cd8+ t细胞的确定细胞群产生的t细胞相比,可表现出至少约1.2倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约4.5倍或至少约5倍更高的倍数扩增。
33.一方面,透过本文所述方法提出的包含至少约50%cd28+ cd8+ t细胞的基因转导t细胞与从包含少于约50%的cd28+ cd8+ t细胞的确定细胞群产生的t细胞相比,可表现出至少约1.2倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约4.5倍或至少约5倍更高的cd8:cd4 t细胞比率。
34.一方面,透过本文所述方法提出的包含至少约50%cd28+ cd8+ t细胞的基因转导t细胞与从包含少于约50%的cd28+ cd8+ t细胞的确定细胞群产生的t细胞相比,可表现出至少约1.2倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约4.5倍或至少约5倍更长的端粒长度。
35.一方面,透过本文所述方法提出的包含至少约50%cd28+ cd8+ t细胞的基因转导t细胞与从包含少于约50%的cd28+ cd8+ t细胞的确定细胞群产生的t细胞相比,可表现出至少约1.2倍、至少约1.5倍、至少约2倍、至少约2.5倍、至少约3倍、至少约3.5倍、至少约4倍、至少约4.5倍或至少约5倍更高的克隆丰富度。
36.另一方面,透过本文所述的方法产生的基因转导t细胞与从包含小于约50%、小于约45%、小于约40%、小于约35%、小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%的cd28+ cd8+ t细胞的确定细胞群产生的t细胞相比,表现出一倍
或多倍更高的倍数扩增、更高的cd8:cd4 t细胞比率、更长的端粒长度和/或更高的克隆丰富度。
37.另一方面,选自包含至少约50%、至少约55%、至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%、至少约91%、至少约92%、至少约93%、至少约94%、至少约95%、至少约96%、至少约97%、至少约98%或至少约99%的cd28+ cd8+ t细胞的确定细胞群的基因转导t细胞与从包含小于约50%、小于约45%、小于约40%、小于约35%、小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%的cd28+ cd8+ t细胞的确定细胞群产生的t细胞相比,表现出一倍或多倍更高的倍数扩增、更高的cd8:cd4 t细胞比率、更长的端粒长度和/或更高的克隆丰富度。
38.另一方面,本公开涉及一种组合物,例如:药物组合物,其包含可透过本文所述方法获得的基因转导t细胞以及药用载体。一方面,本公开涉及治疗患有癌症的患者的方法,包括给予患者透过前述各方面中任一方面中的方法产生的有效治疗量的t细胞,其中所述癌症选自肝细胞癌(hcc)、结直肠癌(crc)、胶质母细胞瘤(gb)、胃癌(gc)、食道癌、非小细胞肺癌(nsclc)、胰腺癌(pc)、肾细胞癌(rcc)、良性前列腺增生(bph)、前列腺癌(pca)、卵巢癌(oc)、黑色素瘤、乳腺癌、慢性淋巴细胞白血病(cll)、梅克尔细胞癌(mcc)、小细胞肺癌(sclc)、非霍奇金淋巴瘤(nhl)、急性骨髓性白血病(aml)、胆囊癌和胆管癌(gbc、ccc)、膀胱癌(ubc)、急性淋巴细胞白血病(all)、多发性骨髓瘤(mm)和子宫癌(uec)组成的组。
39.另一方面,本公开涉及一种用作药剂的组合物,例如:药物组合物,其包含可透过前述各方面中任一方面中的方法获得的基因转导t细胞。
40.另一方面,本公开涉及一种用于治疗癌症的组合物,例如:药物组合物,其包含可透过前述各方面中任一方面中的方法获得的转导t细胞,其中所述癌症选自肝细胞癌(hcc)、结直肠癌(crc)、胶质母细胞瘤(gb)、胃癌(gc)、食道癌、非小细胞肺癌(nsclc)、胰腺癌(pc)、肾细胞癌(rcc)、良性前列腺增生(bph)、前列腺癌(pca)、卵巢癌(oc)、黑色素瘤、乳腺癌、慢性淋巴细胞白血病(cll)、梅克尔细胞癌(mcc)、小细胞肺癌(sclc)、非霍奇金淋巴瘤(nhl)、急性骨髓性白血病(aml)、胆囊癌和胆管癌(gbc、ccc)、膀胱癌(ubc)、急性淋巴细胞白血病(all)、多发性骨髓瘤(mm)和子宫癌(uec)组成的组。
41.另一方面,本公开涉及一种组合物(例如:药物组合物,其包含可透过前述各方面中任一方面中的方法获得的转导t细胞)在治疗癌症中的用途,其中所述癌症选自肝细胞癌(hcc)、结直肠癌(crc)、胶质母细胞瘤(gb)、胃癌(gc)、食道癌、非小细胞肺癌(nsclc)、胰腺癌(pc)、肾细胞癌(rcc)、良性前列腺增生(bph)、前列腺癌(pca)、卵巢癌(oc)、黑色素瘤、乳腺癌、慢性淋巴细胞白血病(cll)、梅克尔细胞癌(mcc)、小细胞肺癌(sclc)、非霍奇金淋巴瘤(nhl)、急性骨髓性白血病(aml)、胆囊癌和胆管癌(gbc、ccc)、膀胱癌(ubc)、急性淋巴细胞白血病(all)、多发性骨髓瘤(mm)和子宫癌(uec)组成的组。
42.另一方面,本公开涉及一种治疗癌症患者的方法,包括从患者中获得cd8+ t胞群、确定所得细胞群中cd28+ cd8+ t细胞的百分比、用抗cd3抗体和抗cd28抗体激活确定的细胞群(前提是确定的细胞群包含至少约50%的cd28+ cd8+ t细胞)或在没有抗cd28抗体的情况下用抗cd3抗体激活确定的细胞群(前提是确定的细胞群包含少于约50%的cd28+ cd8+ t细胞)、用病毒载体转导激活的t细胞群、扩增转导的t细胞群以及给予患者扩增的t细胞
群,其中所述癌症选自肝细胞癌(hcc)、结直肠癌(crc)、胶质母细胞瘤(gb)、胃癌(gc)、食道癌、非小细胞肺癌(nsclc)、胰腺癌(pc)、肾细胞癌(rcc)、良性前列腺增生(bph)、前列腺癌(pca)、卵巢癌(oc)、黑色素瘤、乳腺癌、慢性淋巴细胞白血病(cll)、梅克尔细胞癌(mcc)、小细胞肺癌(sclc)、非霍奇金淋巴瘤(nhl)、急性骨髓性白血病(aml)、胆囊癌和胆管癌(gbc、ccc)、膀胱癌(ubc)、急性淋巴细胞白血病(all)、多发性骨髓瘤(mm)和子宫癌(uec)组成的组。
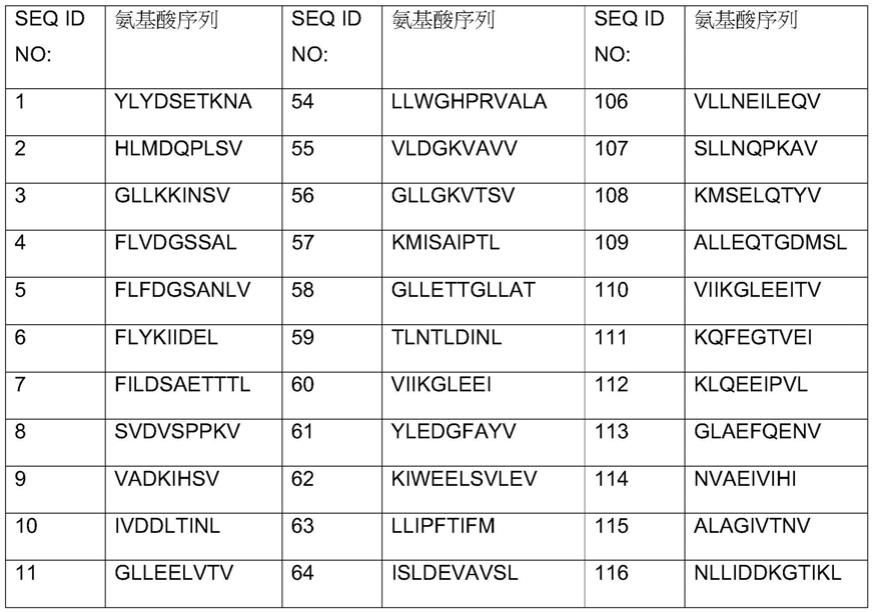
43.另一方面,本公开涉及与和主要组织相容性复合物(mhc)分子复合的肽相结合的tcr,其中所述肽包含选自由seq id no:1
‑
158组成组的氨基酸序列。
44.另一方面,病毒载体可能是表达嵌合抗原受体(car)的逆转录病毒载体。
45.另一方面,病毒载体可能是表达car的慢病毒载体。
46.另一方面,car可能为cd19 car。
47.另一方面,本公开涉及一种治疗癌症患者的方法,包括从患者中获得cd8+ t细胞群、确定所得细胞群中cd28+ cd8+ t细胞的百分比、用抗cd3抗体和抗cd28抗体激活确定的细胞群(前提是确定的细胞群包含至少约50%的cd28+ cd8+ t细胞)或在没有抗cd28抗体的情况下用抗cd3抗体激活确定的细胞群(前提是确定的细胞群包含少于约50%的cd28+ cd8+ t细胞)、用病毒载体转导激活的t细胞群、扩增转导的t细胞群、确定扩增t细胞群的倍数扩增、给予患者扩增的t细胞群(前提是倍数扩增大于10倍),其中所述癌症选自肝细胞癌(hcc)、结直肠癌(crc)、胶质母细胞瘤(gb)、胃癌(gc)、食道癌、非小细胞肺癌(nsclc)、胰腺癌(pc)、肾细胞癌(rcc)、良性前列腺增生(bph)、前列腺癌(pca)、卵巢癌(oc)、黑色素瘤、乳腺癌、慢性淋巴细胞白血病(cll)、梅克尔细胞癌(mcc)、小细胞肺癌(sclc)、非霍奇金淋巴瘤(nhl)、急性骨髓性白血病(aml)、胆囊癌和胆管癌(gbc、ccc)、膀胱癌(ubc)、急性淋巴细胞白血病(all)、多发性骨髓瘤(mm)和子宫癌(uec)组成的组。
48.另一方面,倍数扩增可以为约2至约50倍、约5至约50倍、约10至约50、约2至约30倍、约10至约20倍、约2至约25倍、约5至约25倍、约7至约20倍、约2至约10倍、约2至约5倍。另一方面,倍数扩增可以为大于2倍、大于3倍、大于4倍、大于5倍、大于8倍、大于10倍或大于20倍。附图简要说明
49.为了进一步理解本公开的本质、目的和优点,应参考以下详细描述并结合以下附图,其中类似的元件符号表示类似的元件。
50.图1a显示了根据本公开一实施方案在较大年龄跨度内健康人pbmc的cd8区室中cd28表达百分比。供体的cd28表达透过流式细胞术进行了分析。根据graphpad prism 7中线性回归法确定,观察到cd8 t细胞中起始cd28表达之间呈线性相关性(r2=0.7124)。
51.图1b显示了根据本公开一实施方案在7天制造结束时,cd3区室(即,cd8阳性和cd4阳性区室)内cd8阳性细胞的最终百分比。起始cd28百分比和最终cd8百分比透过流式细胞术计算。根据graphpad prism 7中线性回归法确定,起始cd28百分比和最终cd8百分比之间的r2相关性为0.8121。
52.图1c显示了根据本公开一实施方案7天完成的倍数扩增。起始cd28百分比透过流式细胞术计算。计算转导日至培养期第7天的总倍数扩增。根据graphpad prism 7中线性回归法确定,起始cd28百分比和最终倍数扩增之间的r2相关性为0.8579。
53.图1d显示了根据本公开一实施方案透过流式细胞术测得的最终端粒长度。根据graphpad prism 7中线性回归法确定,起始cd28百分比和最终端粒长度之间的r2相关性为0.9581。
54.图2显示了根据本公开一实施方案所述之t细胞扩增动力学的特征分析。在3名健康供体中,与pbmc的cd8区室中cd28表达中等(mid)(例如54.3%)和较低(low)(例如31.1%)的供体相比,pbmc的cd8区室中cd28表达较高(hi)(例如93.4%)的供体包含更多可以进行早期扩增的t细胞克隆物,如第4天的细胞数相较于第2天的细胞数所定义(用cd3/cd28激活后2天)。
55.图3显示了根据本公开一实施方案,克隆物的收缩和扩增与起始cd28百分比相关。在3名健康供体中,进行了单分子dna测序,并随时间变化追踪了各个t细胞克隆物。差异丰富的百分比表示相对于激活后,截至扩增第10天扩增或收缩可评价t细胞克隆总数的所有t细胞克隆物的部分。cd28表达细胞的百分比根据起始pbmc透过流式细胞术计算。根据graphpad prism 7中线性回归法确定,起始cd28百分比和差异丰富的百分比之间的r2相关性为0.9726。
56.图4显示了根据本公开一实施方案所述之低cd28表达供体表现出负值克隆分裂的延迟t细胞扩增。细胞群增长可基于总活细胞进行计算,并可表示倍数增长。克隆分裂计算为log2(克隆倍数扩增),代表获得的中位值,当培养物中克隆频率收缩时,获得负值(即,虚线以下),而当培养物中克隆频率扩增时,则获得正值(即,虚线以上)。所有点均相对于激活后基线,并计算至t细胞扩增过程第4天。
57.图5显示了根据本公开另一实施方案所述之t细胞扩增动力学的特征分析。根据每个t细胞克隆的log2(倍数增长)所估计已经历的分裂次数,对t细胞克隆物进行分组。早期、中期和后期扩增分别对应于制造过程的第4、7和10天。图例包含当时的中位(med)和平均(avg)克隆分裂以及细胞总数(tot)。
58.图6显示了根据本公开另一实施方案所述之t细胞扩增动力学的特征分析。达到1亿个细胞所需的分裂次数按后期扩增时间点根据平均分裂进行计算。
59.图7显示了根据本公开另一实施方案所述之t细胞扩增动力学的特征分析。计算经历正值或负值早期扩增(第2天至第4天)的t细胞克隆之间的平均最终克隆分裂次数。*p<0.05,**p<.0001。
60.图8显示了根据本公开另一实施方案所述之t细胞扩增动力学的特征分析。刺激后独特克隆爆发后,较低cd28(例如,中等cd28+和低cd28+)的供体中独特克隆不断减少。独特t细胞克隆可源自t细胞受体(tcr)cdr3区独特dna分子读数的数量。数值为1的虚线表示t细胞克隆数少于激活后存在数的点。在t细胞制造过程中,分别在早期(第4天)、中期(第7天)和后期(第10天)测量克隆多样性(独特克隆的数量)。所有值均标准化为在激活后(第2天)时间点独特t细胞克隆的数量。详细说明
61.使用基因改造的t细胞进行过继性t细胞疗法已经成为几种恶性肿瘤的潜在治疗方案。细胞疗法产生的核心是使用刺激、基因工程和扩增方法学组合进行的制造。在此框架内,细胞扩增至治疗相关剂量与保持“活药物”增殖可能性的需求之间可能存在一种微妙的平衡。
62.如本文所述,本公开提出了改善t细胞疗效的方法以及增强和预测t细胞产物的最终倍数扩增、cd8:cd4 t细胞的比率、相对最终端粒长度和克隆丰富度的方法。本公开还提出了有此需要受试者的癌症治疗方法以及透过本文描述的方法产生的t细胞群。
63.cd28是在t细胞上表达的分子之一,这些分子提供t细胞激活所需的共刺激信号。cd28是b7.1(cd80)和b7.2(cd86)的受体。被toll样受体配体激活时,b7.1表达在抗原呈递细胞(apc)中上调。抗原呈递细胞上的b7.2表达为组成性表达。cd28是在幼稚t细胞上组成性表达的唯一b7受体。除了tcr之外,透过cd28的刺激可以为t细胞提供有效的共刺激信号,以产生各种白介素(特别是il
‑
2和il
‑
6)。
64.当延长扩增t细胞时,尽管存在多种增殖细胞因数,它们仍可能丧失其增殖可能性并在功能上衰老。此外,cd28的表达可能与多个制造指标相关,包括最终t细胞倍数扩增。因此,cd28表达的丧失可能为t细胞扩增带来瓶颈,其中某些t细胞克隆相较于制造过程中的其他克隆可能受到极大的青睐。综合多种相关性,可用临床试验资料的荟萃分析显示,年轻患者似乎对涉及cd28共刺激的t细胞制造反应更好,而老年患者似乎对缺乏cd28共刺激的t细胞制造反应更好。
65.本公开的一方面,cd28阳性cd8+ t细胞的起始百分比可以用作生物标志物,以便能够准确预测最终t细胞产物的1)t细胞扩增倍数,2)cd8:cd4 t细胞的比率(或cd3阳性细胞中cd8阳性细胞百分比),以及3)相对端粒长度。另外,cdr3 dna测序可用于追踪来自供体的具有不同起始cd28表达水平的克隆细胞群。根据这项分析,cd28起始表达水平不同可能会导致在整个t细胞制造过程中克隆扩增动力学存在显著差异。
66.t细胞制造工艺依赖于pbmc衍生t细胞的分离、激活和扩增。激活可透过抗cd3和cd28的固定激动性抗体、然后在细胞因数环境中扩增来完成。制造过程中,可能会追踪产品特征(例如,t细胞扩增倍数和cd8+与cd4+细胞的比率),因为这些因素可能会影响治疗效果并达到最低阈值。因此,可能需要更深入地了解可能影响这些指标并影响临床制造结果的因素。
67.例如,t细胞产物制造过程通常可分为五个步骤:(1)白细胞分离术,分离患者的外周血单核细胞(pbmc),(2)激活,(3)用非病毒或病毒编码tcr/car载体对来自pbmc的t细胞进行基因修饰;(4)扩增t细胞,以产生临床相关剂量;以及(5)输注t细胞之前对患者进行可选性淋巴清除,并将修饰的t细胞输注至患者体内。激活t细胞区室可主要透过使用激动性αcd3抗体(进行或不进行αcd28抗体共刺激)、然后通常在il
‑
2中扩增来实现,尽管il
‑
7+il
‑
15可能会产生幼稚t细胞最终产物。
68.扩增过程中,由于撤回tcr刺激,t细胞可能在增长和收缩之间达到平衡。制造过程中,t细胞向终末分化效应细胞进行分化,这一过程可能取决于pbmc的起始分化状态。来自老年供体的pbmc可能富含cd28阴性cd8+ t细胞。此外,基于非细胞凋亡外在fas的t细胞与t细胞相互作用可能会驱动幼稚t细胞的分化。这些观察结果提示,t细胞离体扩增过程中,某些t细胞可能竞争赢过其他t细胞。因此,可能需要在克隆水平上阐明这种收缩和扩增的动力学。
69.在某些方面,本公开的t细胞可能包括原代人t细胞,例如:源自人外周血单核细胞(pbmc)、用g
‑
csf刺激后收集pbmc、骨髓或脐带血的t细胞。条件可能包括使用mrna和dna以及电穿孔。转染后,可能立即输注细胞或者可能储存细胞。在某些方面,转染后,细胞可能在
基因转移入细胞后约1、约2、约3、约4、约5天或更长时间内离体繁殖数天、数周或数月作为主体群。
70.另一方面,转染后,可能克隆转染子,并可能离体扩增证明存在单个整合或附加体型维持的表达盒或质粒且表达tcr的克隆。选择用于扩增的克隆可能证明特异性识别和裂解肽表达靶细胞的能力。可透过用与共同γ链结合的il
‑
2或其他细胞因数(例如:il
‑
7、il
‑
10、il
‑
12、il
‑
15、il
‑
21等)刺激来扩增重组t细胞。可透过用人工抗原呈递细胞刺激来扩增重组t细胞。重组t细胞可在人工抗原呈递细胞上进行扩增,或者用抗体(如:okt3,其与t细胞表面上的cd3交联)进行扩增。重组t细胞亚群可在人工抗原呈递细胞上进行删除,或者用抗体(如:campath,其与t细胞表面上的cd52结合)进行删除。另一方面,可冷冻保存基因修饰的细胞。
71.术语“激活”系指t细胞已被充分刺激而诱导可检测细胞增殖的状态。在特定的实施方案中,激活还可与诱导细胞因数产生和可检测的效应子功能相关。术语“激活的t细胞”尤其系指正在增殖的t细胞。仅透过tcr产生的信号不足以完全激活t细胞,还需要一种或多种次级信号或共刺激信号。因此,t细胞激活包括透过tcr/cd3复合体的初级刺激信号以及一种或多种次级共刺激信号。共刺激可以透过已经接受主要激活信号的t细胞的增殖和/或细胞因数产生来证明,例如透过cd3/tcr复合体或透过cd2的刺激。
72.在某些方面,本公开可能包括制备和/或扩增抗原特异性重定向t细胞的方法,其包括用含有编码tcr的dna构建体的表达载体来转染t细胞,然后,任选地用抗原阳性细胞、重组抗原或受体抗体来刺激细胞,而使细胞增殖。
73.另一方面,提出了使用裸dna或rna透过电穿孔或其他非病毒基因转移(例如但不限于声孔作用)来稳定地转染和重定向t细胞的方法。大多数研究者已使用病毒载体将异源基因携带入t细胞中。透过使用裸dna或rna,可以减少产生重定向t细胞所需的时间。“裸dna或rna”系指以适当表达方向包含于表达盒或载体中的编码tcr的dna或rna。本公开的电穿孔方法产生稳定的转染子,其在其表面上表达并携带tcr。
74.在某些方面,tcr构建体可以作为裸dna或在合适的载体中引入受试者自身t细胞中。本领域中使用裸dna透过电穿孔稳定转染t细胞的方法,请参见,例如,美国专利号6,410,319,其内容透过引用整体并入。裸dna通常系指以适当表达方向包含于质粒表达载体中的编码本公开tcr的dna。有利的情况是,使用裸dna减少产生表达本公开tcr的t细胞所需的时间。
75.或者,可以使用病毒载体(例如,逆转录病毒载体、腺病毒载体、腺相关病毒载体或慢病毒载体)将tcr构建体引入t细胞。根据本公开的方法使用的合适载体在受试者的t细胞中为非复制型。已知有大量基于病毒的载体,其中细胞中维持的病毒拷贝数足够低,可维持细胞的存活性。示例性载体包括:pfb
‑
neo载体以及基于hiv、sv40、ebv、hsv或bpv的载体。
76.一旦确立转染或转导t细胞能够将tcr构建体表达为具有所需调节和所需水平的表面膜蛋白,则可以确定tcr在宿主细胞中是否具有提供所需信号诱导的功能。随后,将转导的t细胞重新引入或给予受试者以激活受试者的抗肿瘤反应。
77.为了便于用药,可以将根据本公开所述的转导t细胞制成药物组合物或制成适于体内给药的植入体,同时具有合适的载剂或稀释剂,其还可以为药用型的。本领域已经描述
了制备这种组合物或植入物的方法(参见,例如,remington's pharmaceutical sciences,16th ed.,mack,ed.(1980年,其内容透过引用整体并入本文))。在合适的时候,可按照其各自的给药途径以常规方法将转导t细胞配制成半固体或液体形式的制剂,例如:胶囊、溶液、注射剂、吸入剂或气雾剂。可利用本领域已知的方法来防止或最小化组合物到达靶组织或器官之前的释放和吸收,或确保组合物定时释放。但是,理想的情况是,使用不妨碍细胞表达tcr的药用形式。因此,理想的情况是,转导t细胞可以制成含有平衡盐溶液(优选为hanks平衡盐溶液)或生理盐水的药物组合物。
78.本公开的方法可用于扩增选择的t细胞群以用于治疗传染病或癌症。得到的t细胞群可以进行基因转导并用于免疫疗法,或者可用于进行感染因子的体外分析。在t细胞群扩增至足够数量后,可将扩增的t细胞恢复至个体。本公开的方法还可提供可再生的t细胞来源。因此,来自个体的t细胞可以离体扩增,一部分扩增的细胞群可重新给予个体,而另一部分则可等分冷冻长期保存,随后扩增和给予个体。类似地,可从患有癌症的个体中获得肿瘤浸润淋巴细胞群,并刺激t细胞增殖至足够数量并恢复至个体。
79.一方面,t细胞的扩增和/或激活在存在一种或多种il
‑
2、il
‑
7、il
‑
10、il
‑
12、il
‑
15、il
‑
21的情况下发生。另一方面,t细胞的扩增和/或激活于单独的il
‑
2、单独的il
‑
7、单独的il
‑
15、il
‑
2和il
‑
15组合或il
‑
7和il
‑
15组合的情况下发生。
80.本公开还可能涉及的组合物含有向t细胞提供共刺激信号以用于t细胞扩增的制剂(例如,抗cd28抗体、b7
‑
1或b7
‑
2配体),其与固相表面偶联,该组合物可能另外包括与相同的固相表面偶联的向t细胞提供主要激活信号的制剂(例如,抗cd3抗体)。这些制剂可能优选附着于珠或烧瓶或袋上。包含每种制剂与不同固相表面偶联(即,提供主要t细胞激活信号的制剂与第一固相表面偶联和提供共刺激信号的制剂与第二固相表面偶联)的组合物也可能在本公开的范围内。
81.本发明的组合物的形式可以为单位剂型,其中每个剂量单位(例如:注射剂)可能含有预定量的组合物,其单独或与其他活性剂适当组合使用。本文所用的单位剂型术语系指适合作为人和动物受试者单位剂量的物理离散单位,每个单位单独含有或与其他活性剂组合含有预定量的本发明组合物,该预定量经计算为足以产生所需效果的量,适当情况下与药用稀释剂、载剂(carrier或vehicle)相关。本公开新单位剂型的规格取决于特定受试者中与药物组合物相关的特定药效学。
82.理想的情况是,有效量或足够量的分离转导t细胞存在于组合物中并引入受试者中,从而可能确立长期、特异性抗肿瘤反应,相较于这种治疗不存在将导致的情况降低肿瘤大小或消除肿瘤生长或再生长。理想的情况是,与除了不存在转导t细胞以外相同的条件相比,重新引入受试者的转导t细胞数量可能使肿瘤大小降低约10%、约20%、约30%、约40%、约50%、约60%、约70%、约80%、约90%、约95%、约98%或约99%。
83.因此,转导t细胞的给予量应考虑给予途径,并且应该使得引入足够数量的转导t细胞以实现所需的治疗反应。此外,本文所述组合物中所含每种活性剂的量(例如,依照每种待接触细胞的量或依照特定体重的量)在不同应用中可能不同。一般来说,转导t细胞的理想浓度应该足以在接受治疗的受试者中提供至少约1
×
106至约1
×
109个转导t细胞/患者m2(或kg),更理想的情况是,约1
×
107至约5
×
108个转导t细胞/患者m2(或kg),但是可使用任何超出,例如,大于5
×
108个细胞/患者m2(或kg),或低于,例如,小于1
×
107个细胞/患者
m2(或kg)的合适的量。给药方案可以基于公认的基于细胞的疗法(参见,例如,美国专利号4,690,915,其内容透过引用整体并入本文),或者可采用交替连续输注策略。
84.这些值可能为在优化实施本发明的本公开方法后医师所使用的转导t细胞范围提供了一般指导。本文中对这些范围的叙述绝不排除使用更高或更低量的组分,因为这在特定应用中可能是必要的。例如,根据组合物是否与其他药物组合物联合使用,或根据药代动力学、药物分布和代谢的个体间差异,实际剂量和方案可能会不同。本领域技术人员可根据特定情况的紧急需要进行任何必要的调整。
85.一方面,能够与本文所述的方法和实施方案一起使用的肿瘤相关抗原(taa)肽包括,例如,美国专利公开号20160187351、美国专利公开号20170165335、美国专利公开号20170035807、美国专利公开号20160280759、美国专利公开号20160287687、美国专利公开号20160346371、美国专利公开号20160368965、美国专利公开号20170022251、美国专利公开号20170002055、美国专利公开号20170029486、美国专利公开号20170037089、美国专利公开号20170136108、美国专利公开号20170101473、美国专利公开号20170096461、美国专利公开号20170165337、美国专利公开号20170189505、美国专利公开号20170173132、美国专利公开号20170296640、美国专利公开号20170253633、美国专利公开号20170260249、美国专利公开号20180051080和美国专利公开号20180164315所述的taa肽,本文所述的这些专利公开内容和序列表透过引用整体并入本文。
86.一方面,本文所述的t细胞选择性地识别呈递上述一个或多个专利和公开内容中所述taa肽的细胞。
87.另一方面,能够与本文所述的方法和实施方案一起使用的taa包括选自seq id no:1至seq id no:158的至少一种。一方面,t细胞选择性地识别呈递seq id no:1
‑
158或本文所述任何专利或申请中所述taa肽的细胞。
88.一方面,能够与本文所述方法一起使用的t细胞受体包括,例如,美国专利公开号20170267738、美国专利公开号20170312350、美国专利公开号20180051080、美国专利公开号20180164315、美国专利公开号20180161396、美国专利公开号20180162922、美国专利公开号20180273602、美国专利公开号20190002556、美国专利公开号20180135039中所述的t细胞受体,这些专利公开的各项内容透过引用整体并入本文。
89.透过本文所述方法产生的基因转导t细胞可改善疗效,更特别地,可改善免疫疗法(例如,过继免疫疗法)的疗效,因为本领域技术人员会理解,透过本文所述方法产生的基因转导t细胞与从包含小于约50%、小于约45%、小于约40%、小于约35%、小于约30%、小于约25%、小于约20%、小于约15%、小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%、小于约2%或小于约1%的cd28+ cd8+ t细胞的确定细胞群产生的t细胞相比,表现出一倍或多倍更高的倍数扩增、更高的cd8:cd4 t细胞比
304830)进行重悬,并在4摄氏度下避光染色15
‑
30分钟,例外情况是,在剩余的表面染色之前,ccr7(ccr7 bv41 biolegend 353208)染色在37摄氏度下在没有血清的rpmi中进行。然后用流动缓冲液洗涤细胞并重悬于固定缓冲液中,在4摄氏度下保存直至在bd fortessa或miltenyi macsquant分析仪上采集。
101.端粒长度测定
102.根据制造商的说明(dako/agilent k5327)测定相对端粒长度。简言之,将t细胞以1:1的比例与对照1301肿瘤细胞(4n基因组)混合。然后通透细胞,将端粒pna fitc探针杂交过夜。次日,进行对比碘化丙啶染色以区分完整的细胞,并透过流式细胞术采集细胞。测试细胞的端粒长度计算为与对照1301肿瘤细胞系端粒长度之比。
103.实施例2
104.cd8+ t细胞上的cd28表达作为用il
‑
7和il
‑
15进行离体t细胞扩增的生物标志物。
105.cd8 t细胞中年龄相关的cd28丧失
106.对于供体之间的选择性压力,供体之间可能存在固有异质性。从pbmc中制造t细胞产物依赖于有效激活和扩增抗原特异性溶细胞cd8 t细胞的能力。在此过程中,可能需要用最小剂量追踪细胞的生长。通常可以基于临床试验设计来满足这种需求。cd28阴性cd8+ t细胞在血液中累积可能会使从老年pbmc制造t细胞产品变得复杂。
107.图1a显示,根据cd28特征分析,供体年龄越大,表达cd28的cd8细胞起始百分比就越低,r2相关性为0.7124(透过graphpad prism 7中的线性回归法确定)。这些细胞对同源肽和透过cd3/cd28刺激的增殖潜能可能降低。
108.与t细胞扩增期间使用il
‑
2相比,il
‑
7和il
‑
15可保留t细胞的幼稚性。因此,il
‑
7和il
‑
15可能是用于临床制造的优选方法。此外,cd28阴性cd8+ t细胞与其cd28阳性对应物相比,可能增殖以回应il15。为了比较在存在il
‑
7和il
‑
15的情况下cd28表达如何影响pbmc衍生t细胞的制造,使用临床类似方法制造了从6名健康供体中获得的t细胞。
109.t细胞扩增期间,cd28起始百分比与最终cd8百分比相关
110.由于cd8区室中cd28表达可能与年龄相关,因此依赖于cd28表达的其他制造指标也可能存在偏差。t细胞扩增结束时,可测量cd8与cd4细胞的比率(或cd3阳性细胞中cd8阳性细胞的百分比),这是因为,尽管细胞溶解cd4细胞已被确定,主要还是cd8区室执行肿瘤细胞溶解功能。因此,cd8细胞中的起始cd28表达与细胞的最终百分比之间可能存在相关性。
111.图1b显示,培养第7天(中期扩增)时,cd8+ t细胞区室中cd28表达的起始百分比与cd3阳性细胞中的最终cd8阳性细胞百分比之间存在相关性,r2相关性为0.8121。这些结果表明,cd28表达可作为cd8+ t细胞选择性压力的驱动力。
112.cd28起始百分比与倍数扩增相关
113.临床t细胞扩增方案通常测量终产物的倍数扩增,作为了解已经发生的细胞群倍增次数。
114.图1c显示,截至第7天(中期扩增),在扩增方案中,倍数扩增与起始cd28表达水平之间存在明显的相关性,r2相关性为0.8579。cd8+细胞生长超过cd4+细胞与cd8+ t细胞上cd28表达的起始百分比密切相关。这些结果对制造工艺的开发具有影响,因为这可基于pbmc的起始表型来预测临床扩增是否会成功。
115.端粒长度减少与起始cd28表达相关
116.端粒长度的损失标志着细胞功能失调,因为这样它们会高度分化并最终衰老。cd3+ cd28刺激后,端粒酶的表达可能仅限于cd4或cd8区室的cd28表达细胞。因此,最终相对端粒长度也可对应于细胞的该cd28表达部分。
117.图1d显示,t细胞产物的最终相对端粒长度可能与起始培养物中cd8+ t细胞上cd28表达水平密切相关,并且pbmc中细胞起始cd28百分比与最终相对端粒长度之间的r2相关性为0.9581。该分析使用来自多名健康供体和多名非小细胞肺癌患者的pbmc进行。资料表明,可以在培养开始之前预测基于il
‑
7/il
‑
15的t细胞制造结果,并且该结果可能对过继t细胞制造方案的设计产生重要影响。例如,由于注入细胞治疗产品的持久性可能与癌症患者的临床结局相关,因此注入肿瘤浸润性淋巴细胞(til)临床产品的最终端粒长度可能与t细胞克隆的持久性有关。
118.综上所述,透过测量起始cd8+ t细胞的cd28表达,可以合理地预测用il
‑
7和il
‑
15制造的t细胞产物的最终cd8百分比、倍数扩增和相对端粒长度。请注意,在cd4+t细胞背景下,可能找不到相同的与cd28表达的相关性,与cd8+ t细胞相比,cd4+ t细胞在衰老过程中可能保持较高水平的cd28表达。
119.实施例3
120.cd8+ t细胞上的cd28表达与t细胞克隆的偏差增殖有关
121.起始pbmc中cd28表达增加为t细胞制造过程中带来有利的生长动力学
122.为了分析t细胞扩增过程中克隆细胞群扩增的特征,在制造过程中透过克隆dna测序和每个克隆细胞群内的绝对数目来追踪单个t细胞克隆的扩增动力学。追踪单个克隆时,分别在扩增过程的早期(第4天)、中期(第7天)和后期(第10天)测量t细胞克隆群内的克隆分裂以及t细胞绝对数量。
123.例如,为了透过基于cd28
低
(31.1%)、cd28
中
(54.3%)或cd28
高
(93.4%)表达水平的cd8+ t细胞的cdr3 dna测序来分析克隆t细胞扩增和收缩的动力学特征,pbmc用激动性cd3/cd28抗体刺激过夜,进行空白转导,然后在制造过程中第4天、第7天和第10天时的扩增过程中取样。由于在每个采样点都进行了细胞计数,并且在每个克隆细胞群内都计算了t细胞的数量,因此可使用以下公式计算克隆分裂次数:克隆倍数扩增=(最终克隆数/起始克隆数)每克隆的估计分裂数=log2(克隆倍数扩增)。
124.除了定量某些cd8+ t细胞群的扩增之外,还可能定量克隆细胞群的收缩,这使用基于增殖染料的技术不可能实现。
125.图2显示,起始pbmc
中
cd28
高
(93.4%)具有早期生长优势,近三分之二(63.41%)的t细胞克隆物在制造的激活步骤(第2天)至第4天之间扩增。相反,cd28表达较低的起始细胞群表现出大多数t细胞克隆物在此制造早期阶段收缩的动力学,其中cd28
中
(54.3%)和cd28
低
(31.1%)细胞群分别包含23.74%和1.19%的早期扩增克隆物。也就是说,在早期扩增阶段,分别有76.26%和98.81%的cd28
中
和cd28
低
表达样本收缩。这与这两个细胞群在此阶段的负值倍数扩增相符,而cd28
高
样本显示出正值倍数扩增。因此,透过在克隆水平上分析t细胞制造的特征发现,扩增过程早期t细胞克隆物可能显著收缩,这可能与表达cd28的cd8+ t细胞的起始百分比呈负相关。
126.克隆物的收缩和扩增与起始cd28百分比相关
127.从单一培养物,追踪各个t细胞克隆频率,并与激活后(第2天)时间点进行比较。透过该比较,评估了频率显著上升和下降的克隆物,总和为差异丰富的百分比。
128.图3显示差异丰富的百分比与起始cd28百分比之间密切相关(r2=0.9726)。起始样本中cd28数量越少,变成差异丰富的克隆物百分比就越高。这表明,在某一细胞群中缺乏cd28会为其他克隆的长入创造生态利基,而缺乏cd28会带来可能在t细胞扩增延长方案期间死亡的细胞群。
129.cd28表达较低的供体显示t细胞扩增延迟以及克隆分裂中位值为负数
130.如果t细胞培养中存在cd28瓶颈,则基于表达cd28的细胞起始百分比,预期t细胞群扩增会延迟。同样,如果所有的t细胞克隆都能立即扩增,则在扩增方案早期每个克隆物的分裂数预期就会为正数。
131.图4显示,对于cd28低和中等表达的培养物(例如:cd28
中
和cd28
低
),在激活后(第2天)和扩增第4天之间存在负细胞群增长,这表明,两个时间点之间的细胞数量有收缩并符合瓶颈事件的定义。另外,仅对于cd28
高
表达的培养物(例如:cd28
高
),观察到总体正值克隆分裂,表明在该培养物中高百分比的t细胞克隆能立即分裂。
132.图5显示,追踪克隆细胞群的分裂时,cd28
低
样本在扩增结束时显示出非正态分布的分裂模式,而cd28
中
和cd28
高
细胞群显示出较为正态分布的特征,即,在整个制造过程中克隆分裂呈正态分布,如相似的平均和中位克隆分裂所示。
133.与收缩增加和早期扩增减少一致的是,cd28
低
细胞群可能需要更多次的克隆分裂来达到培养中既定的扩增水平。也就是说,起始cd28表达越低,达到相同数目的t细胞可能需要进行更多次的分裂。
134.图6显示,cd28
低
细胞群需要1.96次克隆分裂才能达到1x 108个细胞的扩增,而对于相同数目的细胞,cd28
中
细胞群需要1.64次克隆分裂,cd28高细胞群仅分裂0.96次。
135.总而言之,该资料显示,高cd28起始细胞群可能进行更有利的t细胞扩增,且特征为t细胞收缩减少和必需的t细胞分裂次数减少。
136.这些结果表明,t细胞群的cd28表达越高,早期t细胞克隆扩增的可能性就越大。另外,每个限定数目的细胞中t细胞分裂次数减少,这表明cd28表达增加可保留t细胞增殖潜能。
137.为了确定某些克隆细胞群的早期扩增是否可以在整个扩增过程中得以维持,对发生正值或负值早期扩增(第2天至第4天)的t细胞克隆物(例如:cd28
高
、cd28
中
和cd28
低
)之间的平均最终克隆分裂次数进行计算。
138.图7显示,在所有t细胞群中,无论cd28表达如何,在扩增过程(第2天至第4天)结束时,早期扩增的克隆物在统计学上来说更可能分裂。
139.cd28表达较低的供体中扩增期间透过dna克隆测序法测得独特的t细胞克隆物减少
140.在制造激活阶段,可能发生激活诱导的细胞死亡(aicd),并且与较老的效应子样细胞相比,年轻、更幼稚样的t细胞增殖潜能可能更高。因此,这些因素可能导致t细胞制造存在瓶颈,例如,从总细胞群中除去t细胞克隆细胞群而让其他细胞群保留在最终产物中。为了研究aicd对t细胞产物的影响,将整个制造过程中的克隆多样性(或丰富度)确定为衡
cd28方法,而45岁以上的患者可能受益于t细胞制造的单用cd3方法。cr=完全缓解,pr=部分缓解,sd=疾病稳定,nr/pd=无缓解/疾病进展,ne=无法评估。每次临床反应都根据原始临床试验分析来定义。
149.可用临床试验资料的本次荟萃分析显示,较年轻患者(例如,45岁以下)似乎对涉及cd28共刺激的t细胞制造反应更好,而较老患者(例如,45岁以上)似乎对缺乏cd28共刺激的t细胞制造反应更好。
150.多发性骨髓瘤临床试验中,临床反应率与离体倍数增长相关
151.多项临床和临床前研究表明,与较老的较高分化t细胞产物相比,表型上更年轻的较低分化t细胞表现优异。基于制造资料(图1和2),cd28
高
表达(较年轻的)起始pbmc可达到更高的离体倍数扩增,并产生表型上较低分化的最终产物。因此,可以透过在同一时期使用更年轻、较低分化的起始pbmc来制造t细胞并对其进行培养以实现更高的倍数扩增,从而获得较低分化的、效力更强的临床产品。
152.表2显示,根据αbcma多发性骨髓瘤car临床试验,当细胞培养物达到大于10倍的离体扩增时,反应率为57%。相比之下,当培养物未达到10倍扩增时,反应率为0%。这些观察结果为翻译相关性和制造中心模型在预测t细胞效力方面提供了进一步的支持。
153.表2
154.2:离体制造指标与多发性骨髓瘤的临床反应相关。来自临床制造的资料与临床反应率合并,并按照制造过程中达到的cd3+细胞倍数扩增进行分类。反应率计算为相对于该
组患者总人数达到pr或cr的患者人数。pr=部分缓解,sd=疾病稳定,cr=完全缓解。
155.本公开的优点可能包括:透过测量起始cd8+ t细胞的cd28表达来预测t细胞产物的最终cd8百分比、倍数扩增和相对端粒长度,以及基于cd28+ cd8+ t细胞群中cd28表达的起始百分比的个性化疗法。另外,本公开的制造可透过可变的制造周期、起始细胞数量、刺激条件和不同的生长培养基实现个性化。这可改善体外制造指标(例如,倍数扩增),且可能与更好的临床结果相关。本公开的细胞疗法制造可能具有高度的患者特异性,特定组基于彼等之起始细胞表型,对制造的反应可能更好或更差。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1