液态水包油型组合物的制作方法
本发明涉及液态水包油型组合物,更详细地说,涉及虽然是液态,但经时的粘度稳定性和乳化稳定性优异的液态水包油型组合物。
背景技术:
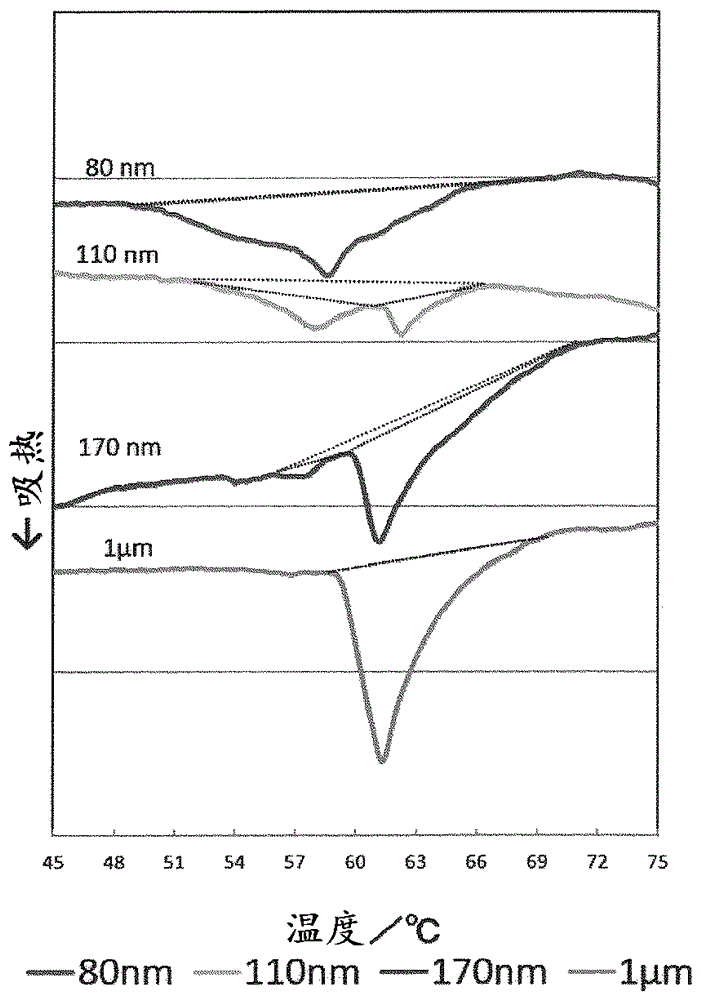
以往,在水包油型组合物中,若提高油剂的含量,则存在经时产生分离或乳析(creaming)等乳化稳定性降低的问题。对此,尝试了以规定的摩尔比掺混亲水性阴离子系表面活性剂和高级脂族醇,形成凝胶转变温度为60℃以上的α凝胶的技术(专利文献1),或者调制由以规定的摩尔比混合高级醇和阴离子型表面活性剂的混合物、和以规定的质量比混合特定的IOB值的水溶性溶剂和水的混合物所构成的双连续微乳液相的α凝胶中间体组合物,向该组合物中添加经加温的油分,接着添加水并进行搅拌,由此诱发α凝胶形成的技术等(专利文献2),利用α凝胶来谋求乳化稳定性的提高。α凝胶是两亲媒性物质形成的自组织体之一,位于固体水合结晶(凝聚胶)和层状液晶的中间,处于在保持结晶状态的同时,还在其亲水基间保持大量的水的状态。凝聚胶那样的几乎不能保持水的结晶是分子自身倾斜而更紧密地填充的状态,相对于此,层状液晶的双分子膜的结构富于流动性,为液体状态,因此可保持大量的水。在α凝胶中,表面活性剂以与层状液晶相同的方式排列成层状,但由于有规则地填充在六方晶中,所以尽管保持了旋转运动,但与层状液晶相比,疏水基的运动性不足,分子的运动受到控制。若通过差示扫描热量分析(DSC)等观测基于这样的结构的运动性的差异,则可观测到作为凝胶-液晶相变的热转移。但是,由于α凝胶是层状液晶与凝聚胶的中间相,所以通常不稳定,通过上述专利文献1和2的技术得到的含有α凝胶的水包油型乳化组合物也存在粘度经时地上升等欠缺粘度稳定性的情况。另外,上述专利文献1和2的技术是基于α凝胶的增粘作用提供乳化稳定性的技术,由于刚制备后就成为较高粘度,所以难以将该技术应用于化妆水等液态的化妆品中。现有技术文献专利文献专利文献1:日本特开2001-348325号公报专利文献2:日本特开2016-14011号公报非专利文献非专利文献1:J.Soc.Cosmet.Chem.Jpn.30(3)310-320(1996),非专利文献2:FoodBiophysics,2012年9月,第7卷第3期,第227-235页,非专利文献3:J.Soc.Cosmet.Chem.Jpn.44(2)103-117(2010)。技术实现要素:发明所要解决的课题本发明的课题在于提供水包油型乳化组合物,其虽然是液态,但经时的粘度稳定性和乳化稳定性优异。解决课题的手段本发明人为了解决上述课题而进行了深入研究,结果发现,通过相对于直链饱和高级醇,以规定量组合N-硬脂酰基-N-甲基牛磺酸钠或N-硬脂酰基-L-谷氨酸及其盐等特定的阴离子型表面活性剂,并且将所形成的乳化滴的粒径调整为160nm以下,不会经时地增粘而可以从制备后起长时间保持液态的性状,并且不会产生乳析或分离等而稳定地维持良好的乳化状态,从而完成了本发明。即,本发明为液态水包油型组合物,其含有以下成分(A)~(C):(A)碳原子数为16以上的直链饱和高级醇,(B)选自N-硬脂酰基-N-甲基牛磺酸钠以及N-硬脂酰基-L-谷氨酸及其盐的1种以上的阴离子型表面活性剂,(C)液态油;且成分(A)相对于成分(B)的含有摩尔比(A/B)为2.8~6,成分(A)和(B)的合计相对于成分(C)的含有质量比((A+B)/C)为0.3~0.75,其中,平均乳化粒径为160nm以下。发明的效果本发明的水包油型乳化组合物虽然为液态,但经时的粘度稳定性和乳化稳定性优异,可长时间不增粘而保持液态的性状,并且不会产生乳析或分离等而稳定地维持良好的乳化状态。另外,在组合物中含有油溶性有效成分的情况下,可提高其经时的稳定性,可稳定地维持基于该有效成分的作用效果。附图说明[图1]表示试验例1中的平均乳化粒径为80nm、110nm、170nm、1μm的组合物的差示扫描热量分析(DSC)的结果的图。[图2]表示试验例1中的平均乳化粒径为80nm、110nm的组合物的X射线衍射结果的图。[图3]试验例1中的平均乳化粒径为110nm的组合物的电子显微镜照片。[图4]参考例1中的脂质体的电子显微镜照片。具体实施方式作为成分(A)碳原子数为16以上的直链饱和高级醇,例如可列举出鲸蜡醇、鲸蜡硬脂醇、硬脂醇、花生醇、山嵛醇、木蜡醇、蜡醇等,可使用它们中的1种或2种以上。其中,从粘度稳定性和乳化稳定性的观点出发,优选鲸蜡硬脂醇、硬脂醇、花生醇、山嵛醇等,特别优选鲸蜡硬脂醇、硬脂醇。从粘度稳定性和乳化稳定性的观点出发,本发明的水包油型组合物中(以下有时简称为“组合物”)的成分(A)的含量优选为0.5~2质量%(以下简称为“%”),更优选为1~1.7%。成分(B)是作为阴离子型表面活性剂的N-硬脂酰基-N-甲基牛磺酸钠、N-硬脂酰基-L-谷氨酸及其盐,作为N-硬脂酰基-L-谷氨酸盐,可列举出钠盐、钾盐、镁盐等,可使用它们中的1种或2种以上。其中,从粘度稳定性和乳化稳定性的观点出发,优选N-硬脂酰基-N-甲基牛磺酸钠、N-硬脂酰基-L-谷氨酸、N-硬脂酰基-L-谷氨酸二钠。从粘度稳定性和乳化稳定性的观点出发,本发明的组合物中的成分(B)的含量优选为0.4~1%,更优选为0.5~0.9%。成分(C)液态油只要是在常温(25℃)下呈液态的油,则可无特别限制地使用,例如可列举出异十二烷、异十六烷、轻质异链烷烃、液体石蜡(矿物油)、角鲨烷、角鲨烯、α-烯烃低聚物、聚丁烯、液体异链烷烃、重质液体异链烷烃、聚异丁烯、氢化聚异丁烯等烃类,油菜籽油、鳄梨油、扁桃仁油、杏核油、苏子油、橙油、橄榄油、奇异果籽油、芝麻油、小麦胚芽油、米胚芽油、米糠油、红花油、鼠尾草油、大豆油、茶籽油、玉米油、菜籽油、月见草油、山茶油、桃仁油(Persicoil)、薏苡仁油、花生油、葵花籽油、葡萄籽油、池花籽油、迷迭香油、希蒙得木油、澳洲坚果油、薰衣草油、玫瑰果油、貂油等动植物油,三(2-乙基己酸)甘油酯、异壬酸异十三烷基酯、异壬酸异壬酯、2-乙基己酸十六烷基酯、肉豆蔻酸异丙酯、棕榈酸异丙酯、棕榈酸-2-乙基己酯、肉豆蔻酸辛基十二烷基酯、三辛酸甘油酯、三(辛酸/癸酸)甘油酯、二异硬脂酸甘油酯、三异硬脂酸甘油酯、十异硬脂酸十甘油酯(十异硬脂酸聚甘油酯-10)、二癸酸丙二醇酯、二癸酸新戊二醇酯、三异硬脂酸聚甘油酯、苹果酸二异硬脂基酯、二乙基己酸新戊二醇酯、季戊四醇四异硬脂酸酯、季戊四醇四(2-乙基己酸)酯、二季戊四醇五异硬脂酸酯、碳酸二烷基酯、双乙氧基二甘醇环己烷-1,4-二羧酸酯、二聚二亚油基氢化松香缩合物等酯类,油酸、异硬脂酸等脂肪酸类,油醇、2-辛基十二醇、2-癸基十四醇、异硬脂醇、2-己基癸醇等高级醇类,二甲基聚硅氧烷(二甲基硅油)、甲基聚三甲基硅氧烷、甲基苯基聚硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、四甲基四氢环四硅氧烷、四甲基四苯基环四硅氧烷、四甲基四(三氟丙基)环四硅氧烷、五甲基五(三氟丙基)环五硅氧烷、聚醚改性甲基聚硅氧烷、油烯基改性甲基聚硅氧烷、聚乙烯吡咯烷酮改性甲基聚硅氧烷等硅油类,全氟聚醚、全氟癸烷、全氟辛烷等氟系油剂类,羊毛脂醋酸酯、羊毛脂脂肪酸异丙酯、羊毛脂醇等羊毛脂衍生物类,对甲氧基肉桂酸2-乙基己酯、水杨酸乙基己酯等液态的紫外线吸收剂等,可使用它们中的1种或2种以上。其中,从粘度稳定性和乳化稳定性的观点出发,优选非极性油,例如可列举出异十二烷、异十六烷、轻质异链烷烃、液体石蜡(矿物油)、角鲨烷、角鲨烯、α-烯烃低聚物、聚丁烯、液体异链烷烃、重质液体异链烷烃、聚异丁烯、氢化聚异丁烯等烃类,二甲基聚硅氧烷(二甲基硅油)、甲基聚三甲基硅氧烷、甲基苯基聚硅氧烷、八甲基环四硅氧烷、十甲基环五硅氧烷、四甲基四氢环四硅氧烷、四甲基四苯基环四硅氧烷、四甲基四(三氟丙基)环四硅氧烷、五甲基五(三氟丙基)环五硅氧烷、聚醚改性甲基聚硅氧烷、油烯基改性甲基聚硅氧烷、聚乙烯吡咯烷酮改性甲基聚硅氧烷等硅油类等,可使用它们中的1种或2种以上。特别适合使用液体石蜡(矿物油)、二甲基聚硅氧烷(二甲基硅油)。从粘度稳定性和乳化稳定性的观点出发,本发明的组合物中的成分(C)的含量优选为1~10%,更优选为1.5~6.5%。另外,从提高油溶性有效成分或粘度、乳化的稳定性的观点出发,极性油相对于非极性油的含有质量比例((极性油)/(非极性油))优选为0.5以下,更优选为0.3以下。需说明的是,在成分(C)液态油中不含后述的成分(D)油溶性有效成分。从粘度稳定性和乳化稳定性的观点出发,本发明中使用的极性油的IOB(无机性一有机性平衡,Inorganic-Organicbalance)更优选为0.05~0.8,进一步更优选为0.05~0.6。从粘度稳定性和乳化稳定性的观点出发,本发明中使用的非极性油的IOB优选为低于0.05,更优选为0.03以下。在这里,本发明中的IOB是用下述(式1)计算的值:IOB=(Σ无机性值/Σ有机性)…(式1)即,IOB可通过如下来计算:基于对各种原子和每个官能团设定的“无机性值”和“有机性值”,将构成表面活性剂等有机化合物的原子和官能团的“无机性值”、“有机性值”积分(参照甲田善生著,“有机概念图-基础与应用-”,11~17页,三共出版,1984年发行)。另外,从粘度稳定性和乳化稳定性的观点出发,液态油的混合IOB优选为0.8以下,更优选为0.6以下,进一步更优选为0.4以下,特别优选为0.2以下。在这里,N种液态油的混合IOB(IOB总)用下述(式2)计算:IBO总=IOB1·W1+IOB2·W2+……IOBN·WN…(式2)IOB1、IOB2、IOBN:各液态油的IOBW1、W2、WN:各液态油的重量分数(W1+W2+……+WN=1)。在本发明的水包油型乳化组合物中,从粘度稳定性和乳化稳定性的观点出发,成分(A)相对于成分(B)的含有摩尔比(A/B)优选为2.8~6,更优选为2.8~4。另外,从粘度稳定性和乳化稳定性的观点出发,成分(A)和(B)的合计相对于成分(C)的含有质量比((A+B)/C))优选为0.3~0.75,更优选为0.3~0.72。从粘度稳定性和乳化稳定性的观点出发,本发明的组合物中的水的含量优选为70~85%,更优选为75~80%。在本发明的组合物中,除了水以外,可使用水性溶剂。作为水性溶剂,例如可列举出乙醇、丙醇等低级醇类,丙二醇、1,3-丁二醇、二丙二醇、1,2-戊二醇、聚乙二醇等二醇类,甘油、双甘油、聚甘油等甘油类等,可使用它们中的1种或2种以上。其中,从粘度稳定性和乳化稳定性的观点出发,优选1,3-丁二醇、二丙二醇、甘油、双甘油、聚甘油。在使用水性溶剂的情况下,从粘度稳定性和乳化稳定性的观点出发,优选将水性溶剂相对于水的含有质量比例(水/水性溶剂)设定为5~20,更优选为6~15。在本发明的组合物中,还可含有成分(D)油溶性有效成分。作为油溶性有效成分,可列举出生育酚、类胡萝卜素、视黄醇、神经酰胺和甘草次酸以及它们的酯,油溶性甘草、油溶性薏苡仁提取物、油溶性迷迭香提取物、泛癸利酮等,可使用它们中的1种或2种以上。作为成分(D),优选选自类胡萝卜素及其酯的1种或2种以上。作为类胡萝卜素,可列举出黄体色素(actinioerythrol)、虾青素、胭脂树橙、角黄素、辣椒质、辣椒玉红素、β-8'-阿朴-胡萝卜素醛(阿朴胡萝卜素醛)、β-12'-阿朴-胡萝卜素醛、α-胡萝卜素、β-胡萝卜素、“胡萝卜素”(α-和β-胡萝卜素类的混合物)、γ-胡萝卜素、δ-胡萝卜素、β-隐黄素、海胆酮、棕榈油胡萝卜素、芦丁、番茄红素、violerythrin、玉米黄质、岩藻黄素、花药黄质、紫黄质等,作为类胡萝卜素的酯,可列举出类胡萝卜素中含有羟基或羧基的物质与脂肪酸或脂肪醇形成了1个或多个酯键的酯等。特别是作为成分(D),优选选自虾青素及其酯的1种或2种以上。形成酯的脂肪酸或脂肪醇优选具有碳原子数为8~24的饱和或不饱和的具有直链或支链的烃链。本发明的组合物中的成分(D)的含量可根据油溶性有效成分的种类适当设定,例如优选为0.00001~1%,更优选为0.00001~0.5。在本发明的组合物中,除了上述成分以外,可根据需要在不损害本发明的效果的范围内使用任意成分。作为任意成分,例如可列举出固体油、粉体、紫外线散射剂、防腐剂、香料等。本发明的组合物可通过将上述必需成分和根据需要掺混的任意成分依据公知的方法乳化混合来调制。例如,将含有成分(A)~成分(C)的油相和含有水的水相分别加热溶解,向水相中添加油相并采用分散等进行乳化混合后,使用微射流机、Ultimaizer、NanoVater等进行高压乳化处理,由此可调制本发明的水包油型组合物。从粘度稳定性和乳化稳定性的观点出发,本发明的水包油型组合物的平均乳化粒径为160nm以下,优选为60~130nm,更优选为70~120nm。平均乳化粒径可通过高压乳化处理的压力、处理时间、处理次数等来调整。在本说明书中平均乳化粒径是通过实施例中记载的方法测定的值。本发明的水包油型组合物通过这样使平均乳化粒径为160nm以下,成为虽然是液态,但经时的粘度稳定性优异的组合物,其理由考虑如下。即,在本发明的水包油型组合物的制备过程中,由成分(A)和(B)形成α凝胶时,若乳化滴的粒径大,则α凝胶的一部分存在于乳化滴的周围而包覆乳化滴,但除此之外的α凝胶处于在连续相中分散地存在的状态。认为这样的剩余的α凝胶经时地形成凝胶的网络而引起粘度上升。相对于此,若使乳化滴的粒径变小,则存在于乳化滴的周围的α凝胶的比例变高,且若平均乳化粒径为160nm以下的范围,则大部分的α凝胶包覆乳化滴,剩余的α凝胶不存在,因此不会产生经时的粘度上升。需说明的是,在使用成分(B)以外的阴离子型表面活性剂或阳离子型表面活性剂的情况下,即使使平均乳化粒径为160nm以下,也不会形成包覆乳化滴的α凝胶膜结构,因此会产生经时的粘度上升。如上所述,在本发明的水包油型组合物中,优选乳化滴的全部或一部分被α凝胶包覆。是否存在这样的包覆乳化滴的α凝胶膜结构(以下有时称为“α凝胶膜结构”)可通过差示扫描热量分析(DSC)测定来确认。即,在乳化滴大的情况(例如平均乳化粒径为1μm左右)下,在DSC测定中在58℃~68℃的范围内可观察到基于剩余的α凝胶的1个吸热峰,但随着乳化滴的粒径变小,在该吸热峰逐渐缩小的同时,可在较低温侧观察到基于包覆乳化滴的α凝胶膜结构的另一吸热峰。这样在DSC测定中,在58~68℃的范围内观察到分别基于剩余α凝胶和α凝胶膜结构的2个吸热峰、或只观察到较低温侧的基于α凝胶膜结构的1个吸热峰的情况下,认为存在包覆乳化滴的α凝胶膜结构。另外,包覆乳化滴的α凝胶膜结构的存在也可通过在X射线衍射中在15nm-1附近观察到峰来确认。即,α凝胶的晶面间距为约4.15Å时,在晶面间距与波数q之间满足q=2×π(圆周率)/d(晶面间距)的式,因此该波数为15nm-1(参照非专利文献1和2、图2)。此外,包覆乳化滴的α凝胶膜结构的存在还可通过电子显微镜观察来确认(参照图3)。认为这样的包覆乳化滴的α凝胶膜结构的存在有助于提高油溶性有效成分的稳定性。即,在水包油型乳化组合物中含有油溶性有效成分的情况下,虽然油溶性有效成分主要存在于乳化滴中,但不会始终停留在相同的位置,而是产生向连续相即水相的分子扩散(奥氏熟化(Ostwaldripening)、参照非专利文献3),因油溶性有效成分与连续相的水等接触而引起油溶性有效成分的劣化。相对于此,推测在本发明中,由于包覆乳化滴的α凝胶膜结构的存在,乳化滴界面变得更坚固,因此可抑制油溶性有效成分的奥氏熟化所伴有的与水等的接触,由此提高油溶性有效成分的经时稳定性。如上所述,在DSC测定中,在58℃~68℃的范围内可包含较高温侧(60℃以上且68℃以下)的基于剩余的α凝胶的吸热峰和较低温侧(58℃以上~低于60℃)的基于包覆乳化滴的α凝胶膜结构的吸热峰中的任一个或两个峰。若将由58℃~68℃的温度范围内所包含的全部吸热峰的面积求得的热量(J/g)计为Q0,将由基于剩余的α凝胶的吸热峰的面积求得的热量计为Q1,将基于包覆乳化滴的α凝胶膜结构的吸热峰的面积求得的热量计为Q2,则从粘度稳定性的观点出发,由基于剩余的α凝胶的吸热峰的面积求得的热量相对于由全部吸热峰的面积求得的热量的比例Q1/Q0优选为0.7以下,更优选为0.2以下,进一步优选为0.1以下。由吸热峰的面积求得的热量Q0、Q1、Q2分别如下求得。即,制作作为得到的DSC曲线的微分值的DDSC曲线,将DDSC曲线开始产生倾斜的温度作为基点。对于DSC曲线,通过将基点与基点连接(基线),可求得峰面积。若将58℃~68℃的温度范围内所包含的全部吸热峰的面积(cm2)计为A0,将基于剩余的α凝胶的吸热峰的面积(cm2)计为A1,将基于包覆乳化滴的α凝胶膜结构的吸热峰的面积(cm2)计为A2,则热量Q0、Q1、Q2可用下式表示:在这里,mi为试样量(g),c为记录数据的进给速度(cm/s),P为DSC曲线的纵轴1cm在1秒钟内相当于几焦耳的值(J/cm·s)。从粘度稳定性和乳化稳定性的观点出发,α凝胶膜结构中的α凝胶膜的膜厚优选为5~20nm,更优选为5~10nm。在本说明书中,α凝胶膜的膜厚指通过电子显微镜观察所测定的100个乳化滴的α凝胶膜的膜厚的平均值。本发明的水包油型乳化组合物可用作化妆品、准药品、药品等,其形态为化妆水、美容液、喷雾剂原液、毛发保护剂、液体粉底等化妆制剂等液态的形态。在本说明书中液态是指,通过实施例中记载的方法测定的粘度为100mPa·s以下,优选为50~0mPa·s。实施例下面列举实施例等,更详细地说明本发明,但本发明不因这些实施例等而受到任何限制。试验例1:由乳化滴的粒径引起的结构变化和稳定性利用下述配方和制法调制平均乳化粒径为80nm、110nm、170nm、1μm的水包油型组合物。对于所得到的各组合物,利用下述条件进行DSC测定。另外,对于平均乳化粒径为80nm、110nm的组合物,通过下述方法进行X射线衍射。此外,对于平均乳化粒径为110nm的组合物,通过下述方法进行电子显微镜观察。将结果示出于图1~3中。(配方)(成分)(%)1.二甲基硅油(25℃6mPa·s)5.02.鲸蜡醇1.53.N-硬脂酰基-N-甲基牛磺酸钠1.04.1,3-丁二醇12.05.尼泊金甲酯0.156.苯氧乙醇0.157.纯化水80.2。(制法)A:将编号1~3均匀地加热混合溶解B:将编号4~7均匀地加热混合溶解C:向B中添加A,乳化混合后冷却D-1:对于C,利用微射流机进行2次高压分散处理(压力为130MPa),调制平均乳化粒径为80nm的水包油型乳化组合物D-2:对于C,利用微射流机进行3次高压分散处理(压力为100MPa),调制平均乳化粒径为110nm的水包油型乳化组合物D-3:对于C,利用微射流机进行2次高压分散处理(压力为80MPa),调制平均乳化粒径为170nm的水包油型乳化组合物D-4:对于C,利用分散器以2000rpm进行搅拌,调制平均乳化粒径为1μm的水包油型乳化组合物。(平均乳化粒径测定方法)测定原理动态散射法溶剂水测定温度20℃测定装置实时纳米粒径测定装置DelsaMaxCORE光源100mWDPSS单纵模激光器激光波长658nm检测器粒径测定雪崩光电二极管(APD)检测器角度粒径测定90°粒径计算方法通过多通道时延(Multi-tau)方式计算自相关系数,并计算粒径。(DSC测定方法)使用设备:EXSTARDSC6200(SeikoInstrumentsInc.制)参比:AIR升温速度:5Cel/min,盘:P/NSSC000E031AL15-CAPSULE分析软件:EXSTAR6000热分析/流变系统(SeikoInstrumentsInc.制)。(X射线衍射方法)测定设备:SAXSess(AntonPaar公司制)测定条件:25℃、20min。如图1所示,在平均乳化粒径为1μm的组合物中,在61℃附近发现由剩余的α凝胶产生的吸热峰。该峰随着乳化滴的粒径变小而缩小,但在平均乳化粒径为110nm的组合物中,除了该峰以外,在58℃附近发现基于包覆乳化滴的α凝胶膜结构的峰。在平均乳化粒径为80nm的组合物中,61℃附近的峰消失,只有58℃附近的峰。由此暗示,若平均乳化粒径为1μm~170nm,则α凝胶主要作为剩余的α凝胶分散在连续相中,但若为110nm,则形成包覆乳化滴的α凝胶与剩余的α凝胶并存的状态,且若为80nm,则α凝胶主要作为包覆乳化滴的物质存在。在平均乳化粒径为110nm、80nm的组合物中,形成包覆乳化滴的α凝胶膜结构也可由X射线衍射中在15nm-1存在峰(图2)和电子显微镜照片(图3)得到证实。基于图1,使用上述分析软件EXSTAR6000求得由全部吸热峰的面积求得的热量Q0、由基于剩余的α凝胶的吸热峰的面积求得的热量Q1和由基于包覆乳化滴的α凝胶膜结构的吸热峰的面积求得的热量Q2,计算由基于剩余的α凝胶的吸热峰的面积求得的热量相对于由全部吸热峰的面积求得的热量的比例Q1/Q0。将结果示出于表1中。[表1]平均乳化粒径Q0Q1Q2Q1/Q080nm1.9101.910110nm1.100.150.330.136170nm1.200.940.040.7831μm1.821.8201对于各组合物,依据下述测定方法和判定标准评价刚制备后和50℃保存1个月后的粘度和乳化状态。将结果示出于表2中。(性状(粘度)的测定/评价方法)在刚制备后和50℃下保管1个月后,测定25℃下的各试样的粘度,依据下述判定标准进行评价。粘度依据使用布鲁克菲尔德(Brookfield)型粘度计的化妆品原料标准·粘度测定法第二法进行测定:(判定):(测定结果)◎:10mPa·s以下〇:超过10mPa·s且为100mPa·s以下×:超过100mPa·s。(性状(乳化状态)的评价方法)对于各试样,通过目视确认刚制备后和50℃下保管1个月后的乳化状态,依据下述判定标准进行评价:(判定):(状态)〇:未发现乳析、分离△:虽然未发现分离,但发现乳析×:发现分离。[表2]由表1和2可确认,与平均乳化粒径为170nm、比例Q1/Q0为0.783的试验例相比,平均乳化粒径为110nm、比例Q1/Q0为0.136的试验例和平均乳化粒径为80nm、比例Q1/Q0为0的试验例即使在50℃保存1个月后也不会增粘,而是维持100mPa·s以下的粘度,经时的粘度稳定性优异。参考例1:含有脂质体的化妆水的调制通过下述制备方法调制如下所示的组成的化妆水。对于所得到的化妆水,进行电子显微镜观察。将电子显微镜照片示出于图4中。参考例1:化妆水(成分)(%)1.磷脂质1.02.黄原胶0.13.羧基乙烯基聚合物0.054.1,3-丁二醇20.05.甘油4.56.氢氧化钠0.027.对羟基苯甲酸酯0.18.香料微量9.纯化水余量。(制备方法)A:使编号1、8和9的一部分脂质体化B:将剩余的成分全部混合C:将A与B混合,得到化妆水。图3中的本发明的具有α凝胶膜结构的乳化滴在内部具有油相,相对于此,图4中的脂质体在内部不具有油相,这是明显不同的。另外,由于脂质体形成多层状膜,可看出膜结构呈多个层状存在,但本发明的α凝胶膜结构观察不到层状的膜结构,可知层状膜数少。另外,可知本发明的α凝胶膜的厚度大约为3分子量的碳原子数为16以上的直链饱和高级醇(α凝胶膜为高级醇3分子膜)。为了形成α凝胶膜结构,除此之外虽然也可以考虑形成5分子膜、7分子膜、9分子膜等,但在那种情况下,有可能因膜数的增加而形成网络结构,从而增粘。因此,从粘度稳定性和乳化稳定性的观点出发,α凝胶膜的膜厚优选为5~20nm,更优选为5~10nm,进一步优选为大约3分子量的碳原子数为16以上的直链饱和高级醇(α凝胶膜为高级醇3分子膜)。实施例1~14和比较例1~12通过下述制备方法调制下述表3~6所示的组成的化妆水。对于所得到的化妆水,依据下述测定方法和判定标准评价刚制备后和50℃保存1个月后的粘度和乳化状态。另外,对于包覆乳化滴的α凝胶膜结构的存在,通过下述方法进行评价。将结果合并示于表3~6中。需说明的是,表3~6的组成中的数值指%。[表3]*1NikkolSMT(NikkoChemicalsCo.,Ltd.制)*2AmisoftHA-P(香荣兴业社制)*3NikkolDDP-8(NikkoChemicalsCo.,Ltd.制)。[表4][表5][表6]<制备方法>(实施例1、7、8、10)A:将编号1~15均匀地加热混合溶解B:将编号16~19均匀地加热混合溶解C:向B中添加A,进行乳化混合D:将C冷却,利用微射流机进行2次高压分散处理(压力为200MPa),得到化妆水。(实施例2、11~14)A:将编号1~15均匀地加热混合溶解B:将编号16~19均匀地加热混合溶解C:向B中添加A,进行乳化混合D:将C冷却,利用微射流机进行1次高压分散处理(压力为200MPa),得到化妆水。(实施例3~6、9、比较例1~12)A:将编号1~15均匀地加热混合溶解B:将编号16~19均匀地加热混合溶解C:向B中添加A,进行乳化混合D:将C冷却,利用微射流机进行2次高压分散处理(压力为100MPa),得到化妆水。<评价方法>(包覆乳化滴的α凝胶膜结构的存在)对于各化妆水,利用下述条件通过DSC测定观察吸热峰,依据以下标准进行判定。[DSC条件]使用设备:EXSTARDSC6200(SeikoInstrumentsInc.制)参比:AIR升温速度:5Cel/min盘:P/NSSC000E031AL15-CAPSULE。[判定标准](判定):(状态)◎:只在58℃附近观察到1个峰〇:分别在58℃附近、61℃附近观察到2个峰△:只在61℃附近观察到1个峰×:未观察到峰-:无法测定。对于通过上述DSC测定被判定为◎或〇的试样,通过电子显微镜观察,观察α凝胶膜结构包覆乳化滴的状况(参照图3,图中的圆所包围的部分)。并且进行X射线衍射(测定设备:SAXSess(AntonPaar公司制),测定条件:25℃、20min),结果在15nm-1发现来源于α凝胶膜结构的峰(参照图2)。(性状(粘度)的测定/评价方法)在刚制备后和50℃下保管1个月后,测定25℃下的各试样的粘度,依据下述判定标准进行评价。粘度依据使用布鲁克菲尔德型粘度计的化妆品原料标准·粘度测定法第二法进行测定。(判定):(测定结果)◎:10mPa·s以下〇:超过10mPa·s且为100mPa·s以下×:超过100mPa·s。(性状(乳化状态)的评价方法)对于各试样,通过目视确认刚制备后和50℃下保管1个月后的乳化状态,依据下述判定标准进行评价。(判定):(状态)〇:未发现乳析、分离△:虽然未发现分离,但发现乳析×:发现分离。如表3和4所示,关于实施例1~14的化妆水,均发现包覆乳化滴的α凝胶膜结构的存在,从刚制备后起呈现液态,即使在50℃保存1个月后也未增粘,而是维持100mPa·s以下的粘度。另外,也不会产生分离或乳析,保持良好的乳化状态。相对于此,关于平均乳化粒径为170nm以上的比较例1~2的化妆水,未确认到α凝胶膜结构的存在,发现经时的粘度上升。另外,代替成分(A)碳原子数为16以上的直链饱和高级醇,使用具有分支的物质或碳原子数低于16的物质的比较例3~5的化妆水均从刚制备后就发现分离(初期分离)。在使用非离子型表面活性剂代替成分(B)的情况下也同样地产生初期分离(比较例6),在使用阳离子型表面活性剂或成分(B)以外的阴离子型表面活性剂的情况下,未确认到α凝胶膜结构的存在,即使使平均乳化粒径为160nm以下,在50℃保存1个月后也会分离或经时地粘度上升等,乳化稳定性和粘度稳定性差(比较例11、12)。另外,在成分(C)相对于成分(A)和(B)的合计的含有质量比在0.3~0.75的范围以外的情况下,乳化稳定性或粘度稳定性同样差(比较例7~10)。试验例2:油溶性有效成分的稳定性试验通过下述制备方法调制下述表7所示的组成的化妆水。对于所得到的化妆水,通过下述方法评价油溶性有效成分的稳定性。[表7]*4虾青素-5C(OryzaOil&FatChemicalCo.,Ltd.制)*5β-胡萝卜素(三共制药)*6Kaneka辅酶Q10(Kaneka)。(制备方法)A:将编号1~8均匀地加热混合溶解B:将编号9~12均匀地加热混合溶解C:向B中添加A,进行乳化混合D:将C冷却,利用微射流机进行2次高压分散处理(压力为130MPa),得到化妆水。(虾青素、β-胡萝卜素和泛癸利酮的稳定性评价方法)对于各化妆水,测定刚制备后和50℃下保管1个月后的吸光度。使用分光光度计UV-2500PCUV-VISREDCORDINGSPECTROPHOTOMETER(SHIMADZU公司制),用光路长10mm×光路宽10mm的玻璃比色池,使用纯化水作为参比,虾青素在480nm波长附近、β-胡萝卜素在450nm波长附近、泛癸利酮在280nm附近测定吸光度。通过下式计算吸光度残存率,并依据下述标准评价虾青素、β-胡萝卜素、泛癸利酮的稳定性:吸光度残存率(%)=(50℃下保管1个月后的试样的吸光度)×100/(刚制备后的试样的吸光度)。<稳定性判定标准>(判定):(吸光度残存率)◎:吸光度残存率为60%以上〇:吸光度残存率为50%以上且低于60%×:吸光度残存率低于50%。如表7所示,在实施例15的化妆水中,与比较例13相比,在50℃保存1个月后的虾青素的褪色得到明显地抑制。认为其原因在于,在实施例15中α凝胶膜结构包覆乳化滴,乳化滴界面变得更坚固,因此抑制了虾青素的奥氏熟化所伴有的与水等的接触,由此抑制了虾青素的分解等,相对于此,在比较例13的化妆水中乳化滴的周围未形成α凝胶膜结构,因此产生了虾青素与水等的接触和与之相伴的分解等。对β-胡萝卜素和泛癸利酮也同样,确认在50℃保存1个月后也稳定维持。试验例3:极性油与非极性油之比和稳定性通过下述制备方法调制下述表8所示的组成的化妆水。对于所得到的化妆水,与实施例1~14同样,评价刚制备后和50℃保存1个月后的粘度和乳化状态。另外,对于虾青素的稳定性,与试验例2同样地进行评价。将结果合并示于表6中。[表8]*7HICALLK-230(Kaneda)*8MyritolGTEH(BASF)。(制备方法)A:将编号1~5均匀地加热混合溶解B:将编号6~9均匀地加热混合溶解C:向B中添加A,进行乳化混合D:将C冷却,利用微射流机进行2次高压分散处理(压力为130MPa),得到化妆水。如表8所示,实施例19~21和实施例25的化妆水与实施例27的化妆水相比,在50℃保存1个月后的粘度稳定性和乳化稳定性更好。此外,实施例22~24和实施例26的化妆水与实施例28的化妆水相比,在50℃保存1个月后的粘度稳定性、乳化稳定性和油溶性有效成分的稳定性更好。认为其原因在于,极性油相对于非极性油的含有质量比例((极性油)/(非极性油))越低,与直链饱和高级醇的相容性越下降,α凝胶膜结构的高温经时稳定性越良好。实施例27:化妆水(成分)(%)1.二甲基硅油(25℃6mPa·s)4.52.棕榈酸乙基己酯*90.53.鲸蜡硬脂醇1.54.山嵛醇0.85.鲸蜡硬脂醇0.86.N-硬脂酰基-N-甲基牛磺酸钠0.77.甘油1.08.1,3-丁二醇12.09.三丙二醇0.510.柠檬酸0.0111.柠檬酸Na0.0112.纯化水余量*9SALACOSP-8(NisshinOillioGroup公司制)。(制备方法)A:将编号1~5均匀地加热混合溶解B:将编号6~12均匀地加热混合溶解C:向B中添加A,进行乳化混合D:将C冷却,利用微射流机进行2次高压分散处理(压力为130MPa),得到化妆水。如上所述得到的实施例27的化妆水在刚制备后为30mPa·s的液态,平均乳化粒径为80nm,乳化状态也良好。50℃下经过1个月时的粘度稳定性、乳化稳定性也良好。产业上的可利用性本发明的水包油型乳化组合物虽然是液态,但经时的粘度稳定性和乳化稳定性优异,因此可用于液态的化妆品或准药品等。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1