优化生物序列理化性质的由计算机实现的方法与流程
1.本发明涉及一种由计算机实现的方法,用于优化高分子生物分子(尤其是蛋白质和核酸)的物理化学性质。尤其,本发明涉及一种自动计算方法,用于预测、识别、选择和生成序列,这些序列具有与分子令人感兴趣的理化性质有关的最佳特性。
2.尤其,本发明涉及一种对来源于深度突变扫描(dms)和定向进化(de)实验的数据进行分析和选择的方法。
3.本发明还涉及一种预测生物序列的物理和化学性质的方法,该方法以使用来源于hts-selex(高通量测序selex)实验的数据为基础。
背景技术:
4.深度突变扫描和定向进化方法是研究诱变得到的不同蛋白质变体的影响和准备这些变体的功能性选择的途径。
5.尤其对于所有需要可靠产品和高产量的工业和制药应用而言,能够产生氨基酸序列并且同时预测其具体行为仍然是未解决的挑战。
6.当前,实验室中蛋白质工程的方法分为两个主要类别:体内实验方法,诸如引导进化,或者,计算机模拟(in-silico)方法,诸如合理设计。
7.在第一种情况下,在实验室中重现并加速了生物进化过程(packer and liu-nature reviews genetics-2015);该技术涉及创建参考蛋白质突变体文库和执行渐进循环,包括:选择步骤,其中针对给定功能表达并分离突变的变体,和选择的突变体的进一步诱变步骤。该技术已经成功地应用于化学合成酶的优化。引导进化技术存在基本限制,只能够研究给定序列的大量理论上可能的变体中的一小部分。由于这个原因,通常,被测试的样本是具有明确限定的点突变的样本或重组现有已知序列产生的突变体。
8.相反,合理设计方法是基于结构信息,这些结构信息最初来源于结晶实验或来源于蛋白质可能结构的计算机模拟预测;因此,可能的蛋白质结构和序列的模拟使用与氨基酸置换引起的自由能变化有关的估算来实施,这能够使用得分函数来计算。
9.对于这种方法,在构象研究(通常具有侧链旋转异构体文库)中需要有效的算法,以便举例说明设计的或改性的结构的可能构象。通常,得分函数和构象搜索算法使用天然蛋白质信息或称作起点的接口(interface)。
10.还有一些方法试图使用基于序列的策略来产生功能蛋白。该方法使用机器学习技术来描述特定靶蛋白的同源序列的统计约束(barrat-charlaix et al.-scientific reports)(hopf et al.-nature biotechnology)。然而,有一些最先进的案例,其中,已经有可能成功地设计新的蛋白质(de-novo protein)。bakers和同事(po-ssu huang et al.nature 2016)设法设计并生成了具有自然界中从未描述过的折叠结构的蛋白质并且设法改变了蛋白质-蛋白质相互作用。
11.深度突变扫描(dms)是最近的方法,其结合从大规模诱变实验中选择的数据来调查研究蛋白质功能和dna测序技术的高通量测序,以便对蛋白质的不同变体的活性进行量
化(fowler dm and fields s.-nat methods,2014)。
12.在这种方法中,首先将蛋白质变体文库引入到模式生物,例如,噬菌体、细菌、酵母或哺乳动物细胞培养物。一旦被研究的蛋白质及其不同突变体被表达,就针对预期的蛋白质功能或针对所感兴趣的其它分子性质执行选择步骤;该步骤具有基于功能容量来丰富每个变体频率的功能,该功能容量例如是适应度、结合选择的靶标的能力或催化酶促反应的能力。
13.在每个循环中,使用dna深度测序技术测定每个突变体的频率,以便计算每个变体出现的次数。这些测序技术的问世已经极大地提高了测试蛋白质的序列-功能关系的能力。至今,dms数据分析主要基于归于每个蛋白质变体的富集得分;该得分用于对选择的突变体的偏好进行量化,计算为选择前后突变体频率的比值,并在每一轮中与原始(野生型)蛋白质的得分进行比较。
14.参考软件是enrich2(alan f.rubin et al.genome biology 2017)。该软件基于抽样误差和与实验重复的一致性(如果可用)使用措施来修正得分。在多个循环的情况下,其使用线性回归来评价最终得分。
15.尽管使用了措施,富集得分仍会受到各种性质的统计噪声的影响,例如,因为以下原因产生的噪声:抽样,实验重复的低再现性和时间序列中频率变化的非线性,以及实验对起始文库的强依赖性和实验的其它特性。
16.分析dms数据的另一工具是dms_tools2(jesse d bloom bmc bioinformatics 2015)。dms数据分析中其它令人感兴趣的出版物如下:
17.otwinowski,jakub.“biophysical inference of epistasis and the effects of mutations on protein stability and function.”molecular biology and evolution 35.10(2018):2345-2354。
18.适体(aptamer)是短的rna或dna分子,能够以高亲和力和特异性结合靶分子,在治疗和诊断领域中具有极大的应用潜力。用于选择功能性适体的最常用的一项技术称作selex(指数富集的配体系统进化技术)。
19.经典方法包括合成独特的寡核苷酸序列的文库,该寡核苷酸序列的结构具有中心部位,其含有随机产生的序列,并排地具有两个保留序列的端部。该技术的步骤包括与靶分子结合的几个选择循环、选择的序列的分离和随后的扩增。
20.几个循环结束时,富集了具有较高结合能力的序列,使用深度测序技术测定它们的频率。在这种情况下,我们推荐高通量测序selex(hts-selex)。该技术有几个衍生的技术,但保留了选择和富集大多数功能序列的理念。
21.正如dms方法一样,需要使用非常大量的数据,对于研究序列的富集,有一些不同的分析方法。
22.因此,仍然需要开发有效且可靠的方法用于对筛选实验(dms、de和selex)得到的序列文库进行计算机模拟分析,该方法可选择具有所期望的特性的突变体、预测未经实验测试的突变体的适应度并产生具有优化特性的突变体。尤其,需要预测突变效果的方法,该方法会考虑个体突变的非叠加的上位性效应。
23.shen et al.在
‘
protein engineering,desing and selectio’vol.21no.1of 19december 2007中描述了一种方法,其中,用于模型训练的输入数据需要吉布斯自由能的
实验测定值,测量吉布斯自由能时的温度和ph。这需要时间和费用上的巨大投资来实施这些测量。
技术实现要素:
24.根据本发明的方法已经解决了现有技术的问题。
25.发明人现在已经开发了一种方法,用于分析来源于dms、de和selex实验的数据文库。
26.因此,本发明的目的是提供一种有效且可靠的方法,用于对从这样的实验得到的序列文库进行计算机模拟筛选并且选择出所期望特性的最佳突变体。
27.本发明的另一目的是一种方法,该方法提供了将来源于dms实验、selex的一组序列或序列文库用于产生第二组高效率生物序列的用途,其中,高效率的意思例如是高催化能力、高适应度、与特定靶标结合的能力高、高荧光活性,一般而言,高性能是相对于最开始界定的分子的理化性质而言的,能够通过上述实验进行选择。
28.由基于计算机的方法至少部分地实现了本发明的范围,以便分析生物序列,从而优化生物序列(例如,蛋白质、rna、dna等)的生物物理性质,这与分子的理化特性有关,还包括生物特性,诸如,与特定靶分子(例如,结合特定抗原的抗体)结合、催化作用、荧光性、热稳定性、免疫能力(抗体抗感染活性的ic50)等;该方法包括如下步骤:
[0029]-定义允许的或令人感兴趣的分子状态(例如,分子的宏观状态是结合的、未结合的、折叠的、非折叠的等),这些分子状态与分子的令人感兴趣的活性或理化特性有关;
[0030]-定义与允许的分子状态有关的至少一个函数,下文中是定义的统计能量。不同状态的这些统计能量是通用序列的函数,因此定义基因型到表型的图谱(即,从序列到所考虑的理化活性的图谱)。这些函数依赖于标量参数的多元线性函数,标量参数可能与位置偏好有关,例如包含蛋白质特定位置处氨基酸能量贡献的参数,并且,可能与上位性效应有关,诸如成对及更高水平的相互作用(例如三联体、四联体等),这考虑了突变效应的不可加性。
[0031]-提供描述筛选实验的一个或多个循环(例如,定向进化、深度突变扫描、selex等)的似然函数。该概率函数至少包括第一概率因子,其表示基于至少一个统计能量函数来选择序列的概率。优选地,还存在其它因子,例如,第二概率因子,其表示在实验筛选过程中扩增给定序列的概率。通常,似然函数的概率因子是筛选实验各步骤的概率表达式。尤其,这些概率取决于:a)通过上述统计能量函数定义得到的每一分子状态的模型参数;b)在应用该方法之前进行的实验筛选循环的测序检测的每个变体的样本数目。
[0032]-通过使参数本身的后验概率最大化来测定至少一个统计能量函数的参数,将从一个或多个筛选实验的测序得到的训练数据分配给这些参数,这些筛选实验用于针对期望的理化特性(例如,与不同靶标连接)来选择分子的变体。尤其,对每一可能的序列变体的参数进行计算。
[0033]-基于通过训练阶段得到的参数,计算与所考虑的序列分子状态有关的统计能量函数;
[0034]-基于至少一个统计能量函数,向与期望的理化特性有关的指定序列提供得分。例如,为了预测与指定序列的靶标的亲和力,针对与靶标相关的状态,采用关于序列的统计能量来定义得分。因为每个可能的序列变体的参数都是可用的,所以基于指定序列来计算统
计能量。计算能量参数之后指定的不同序列将对应于不同分值;和/或
[0035]-基于可能的序列的整个集合或针对其特定子集,利用最大化的得分或高于预定阈值的得分来产生序列或序列文库。优选地,应当将该集合理解为长度与训练所用实验序列的长度相等的序列。根据能量参数的该用途,应用优化算法,该算法可发现能够使能量参数最大化或使训练阶段已经计算的能量参数的函数最大化的一个或多个序列。通过该方法,尤其是通过得分的归因分析(attribution),有可能处理大量输入序列,以便针对所期望的理化特性(即,催化作用、适应度、与特定靶标结合的能力、荧光活性、免疫能力等)分析突变体的行为。
[0036]
应当注意的是,通过用于训练模型(即用于计算多元线性函数的参数)的生物样本序列集合来识别所期望的理化特性。该集合包括多个序列,这些序列具有/显示目标理化特性/对目标理化特性有效。
[0037]
以这种方式,针对由本发明的方法指定并处理的每个序列,有可能通过得分由计算机模拟评价该输入序列的相关性有多大,也就是说,它是针对目标理化性质而提供的或者显示了目标理化性质或者对目标理化性质是有效的。
[0038]
此外,对目标理化性质具有高亲和力的序列文库包括在体外或体内连续实验阶段使用的许多潜在有效的蛋白质序列。
[0039]
还应当注意的是,基于训练阶段的输入样本序列来计算多元线性函数的参数。将样本序列用作训练阶段的输入数据(优选唯一类型的输入数据)以便实现无监督训练,这能极大节省成本,因为不需要对参数诸如ph和/或吉布斯温度和/或自由能进行具体测量。根据本发明的方法,通过该方法可以计算吉布斯自由能或吉布斯自由能是可计算的,并且,在定义输入数据的实验中不用测量吉布斯自由能。
[0040]
已经证实,有可能建立通用筛选实验的似然函数,并且,有可能基于一个或多个多元线性统计能量函数采用下面实施例中描述的装置来表达相关因子,该相关因子代表总是存在于这样的实验中的选择阶段,每个多元线性统计能量函数都具有关于所有可能序列的参数。根据具体筛选实验的阶段,本领域技术人员能够建立除选择的概率因子之外的任何概率因子,其适合于完成似然函数。
[0041]
通过使似然函数最大化来计算至少一个统计能量函数的标量参数,这使似然函数可用作计算通用序列得分的基础,这可以有多种表达式,尤其,最简单的是序列的得分是与所述序列有关的标量参数之和。
[0042]
因此,在计算之后,统计能量函数的参数可用于借助于得分来评价一个或多个输入序列,或者,用于生成使能量参数的函数最大化的一个或多个序列。
[0043]
此外,有可能的是:
[0044]-根据与令人感兴趣的每个性质有关的统计能量函数,提供得分,该得分考虑了分子不同生物物理特性。例如,通过使结合能与折叠的能量相结合,有可能定义得分,该得分考虑了分子的稳定性和亲和力;和/或
[0045]-通过选择不同的函数(例如,与不同靶分子结合),提供得分,该得分使用相同分子的多重筛选实验。在这种情况下,本发明的方法提供了与每个实验(即不同函数)有关的序列能量,有可能将这些个性化分数的能量进行结合,用于得到分子生物物理函数的期望组合。例如,这对于设计具有结合能的多特异性变体是有用的,这些结合能分配给筛选中使
用的靶标(例如,双特异性单克隆抗体,或者更一般地说,多特异性抗体);和/或
[0046]
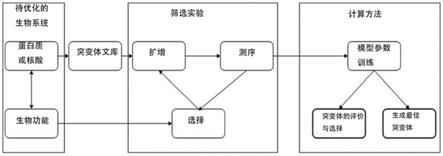
在本发明的一个实施方案中,优化的文库可以用作用于后续实验或用于产生新的随机序列的一组初始序列。借助于流程图和通过该流程实现的突变体文库的生成,可以理解该过程,其中流程图包括筛选实验和模型的计算机模拟训练。
[0047]
其他目的将从下面对本发明的详细描述中显而易见。
附图说明
[0048]
图1
[0049]
与该方法的输入/输出实施例有关的一般流程图。
[0050]
图2
[0051]
与该方法创建途径实施例有关的一般流程图,该方法的创建:从对实验测序的原始数据进行预处理开始到模型输出的三个实施例,这不视为穷尽了其用途。
[0052]
图3
[0053]
模型说明中使用的主要定义的图示。
[0054]
左侧方框:ns(t)是显示序列s的载体(例如噬菌体)的数目。从测序得到的读取数与ns(t)成比例。
[0055]
右侧方框:ns(t)是有序列s的载体的数目,该序列s已被选择(例如,已经与靶标结合)。
[0056]
图4
[0057]
突变体选择性的评价。由模型计算的统计结合能的散点图以及根据数据计算的选择性(选择性公式如下文所示)。图中的四个方框与实施例1描述的实验有关。
[0058]
圆圈(十字)是测试集(训练集)上的序列,初始计数超出某阈值(检查数据质量的程序)。每个方框中,每种情况都有斯皮尔曼相关系数(spearman correlation coefficient)。斯皮尔曼系数用于衡量两个变量之间的顺序关系的程度。强相关性,系数值介于0.80至0.98,显示了在四个实验数据集的每一个中模型预测结合亲和力的能力。
[0059]
图5
[0060]
突变体选择性的评价。由模型计算的结合能分散图以及根据数据计算的选择性。
[0061]
该图形显示了通过在与同一蛋白质相关的另一个实验(实施例3)上学习模型来评价实验(实施例2)序列的选择性。
[0062]
因此,点对应于属于实验2的序列,总计数超出某阈值(检查数据质量的程序)。横坐标显示由实验2计数计算的选择性,纵坐标表示由实验3数据初始化的模型的统计能。
[0063]
强相关性,皮尔逊系数(pearson's coefficient),显示了在不依赖于学习的实验中模型预测结合亲和力的能力。两个统计变量之间的皮尔逊系数表示它们之间任何线性关系的程度。
[0064]
图6
[0065]
突变体选择性的评价。由模型计算的键能的散点图以及根据数据计算的选择性。
[0066]
该图显示了通过学习低选择性序列的模型来评价高选择性(在这种情况下是指高结合亲和力)序列的选择性。数据与实验1相关。应注意,在该测试中,在学习或初始化阶段,具有高亲和力且因此先验信息更丰富的序列隐藏于模型中。
[0067]
黑点对应于低选择性训练序列,而灰色十字对应于高选择性测试序列。
[0068]
再次,模型分数根据实验选择性对序列进行正确排序
具体实施方式
[0069]
下面列出的定义用于本发明的描述中。
[0070]
深度突变扫描(dms)是一种基于下一代测序技术的方法,用于测量一些独特变体的活性,在单一实验中大约是105(或更多)个蛋白质、dna序列或参考rna。
[0071]
selex(指数富集的配体系统进化)是一种组合生化技术,适合于产生dna或rna寡核苷酸(单链和双链),这些dna或rna寡核苷酸能够特异性结合称为适体的给定靶标。
[0072]
定向进化或de是一项技术,在该技术中,由一个或多个序列构建变体文库,针对感兴趣的性质对变体文库进行选择,最佳变体或选择的变体用于下一个循环(round),在下一个循环中重复进行该流程。
[0073]
机器学习辅助定向进化的意思是直接进化过程,在该过程中,从序列选择数据开始(对所选序列的样本进行测序)在给定的或多个循环中训练计算机模拟模型,该计算机模拟模型用于向下一个循环推荐变体,如"machine learning-assisted directed protein evolution with combinatorial libraries",z.wu et al.arxiv preprint中所描述。
[0074]
深度测序是指dna给定区域的重复测序技术(数百次或上千次)。该新一代测序方法能够检测稀有的克隆类型或微生物细胞,它们的遗传贡献约为所分析遗传物质的1%。
[0075]
超深测序技术是指一种仅限于基因组有限区域的特殊深度测序技术,能够检测遗传贡献百分比可能是大约10-7
/10-8
的变体。
[0076]
基因突变是指由于外部因素或偶然因素,但不是由于基因重组,在基因组或更普遍的遗传物质(dna和rna)的核苷酸序列中发生的任何稳定和可遗传的改变。
[0077]
体细胞突变是指非遗传基因突变。
[0078]
折叠或蛋白质折叠是指蛋白质达到其三维结构的分子折叠过程。相反,蛋白质的非折叠状态是指线性多肽链的变性状态。
[0079]
表型是指一个活生物体所表现出的所有特征的集合,涉及其形态学、发育学、生物化学和生理特性,包括行为。从广义上讲,我们指的是基因组编码区中一个或多个突变的表型,即功能或结构变异。
[0080]
基因型是指构成生物体或种群dna(遗传组成/遗传特性/遗传构成)的所有基因的集合。
[0081]
上位性(epistasis)通常是指个体突变之间的非加性表型效应。
[0082]
残基是指蛋白质或多肽的氨基酸。
[0083]
分子状态是指生物分子的状态(例如,结合态、非结合态、折叠态、非折叠态等),与其活性或理化特性相关。
[0084]
编码序列是指基因中编码蛋白质的dna或rna部分。
[0085]
序列比对是指生物信息学程序,在该程序中,通过插入合适的插入和缺失标记(不是与氨基酸或含氮碱基相关的描述标记),将氨基酸、dna或rna的两个或更多个一级序列排列在共长矩阵中。
[0086]
位置偏好(positional bias)是指在给定序列文库的特定位置或更一般地在多序
z,et al.-recent advances in selex technology and aptamer applications in biomedicine.int j mol sci.2017oct 14;18(10)”,已经开发出各种变体。
[0099]
因此,根据本发明的方法是以使用模型为基础,该模型能够评价相对于训练集序列呈现一个或多个突变的序列,不仅考虑这些突变可能沿序列在不同位置发生的概率,而且根据实施例,还考虑它们的相对上位性。
[0100]
通过基于方法描述部分中序列的统计能量函数的得分进行选择。
[0101]
一旦模型在一组输入序列上已经进行了训练(例如,dms实验),该模型可用于评价与实验筛选中使用的突变文库匹配的任何序列。
[0102]
概率模型的描述
[0103]
在一种优选的实施方案中,下面描述的模型考虑了两种状态,选择的或未选择的,借助于这些状态,每个序列都有一个概率。
[0104]
然而,可归纳成几种状态,借助于这些状态,每个序列都有一个概率。例如,在一种优选的实施方案中,考虑三种状态:结合的、非结合并折叠的、非折叠的。
[0105]
在对模型的详细描述中,参考通用版本,该通用版本考虑了一般数量的状态,尤其是,对两种状态的情况进行了描述。
[0106]
标记
[0107]
下划线符号x是指载体,其要素表示序列{xs}。粗体符号x表示每个序列s和重复t的在所有序列和循环的一组分布量,{xs(t)}。
[0108]
符号的定义
[0109]rs
(t)是循环t中序列s的读取数
[0110]ns
(t)是循环t中运输序列s的载体数目
[0111]
是循环t中载体的总数
[0112]
是循环t中读取的总数
[0113]
是循环t中载体的总数
[0114]ns,k
(t)是循环t中载体的数目,这些载体具有处于状态k的序列s
[0115]
d是不同序列的数目
[0116]ek
(s)是处于状态k的序列s的统计能量
[0117]
k∈sel是分子离散状态的集合,用于指定接下来的一轮筛选实验的选择,训练数据来自于该筛选实验(例如,非特异性结合、结合、折叠、非折叠)。
[0118]
通常,统计能量是线性多元函数。在第一个实施例中,将每种状态的能量定义为能量参数的线性函数,将独立的位置偏好与上位性效应(即突变效应的不可加性)表示为二重、三重相互作用等:
[0119][0120]
每个是取决于具有位置i1,...,i
p
的p个氨基酸的统计能量
贡献。总之,它们构建了训练过程中计算为标量的自由参数。
[0121]
上述表达式不应被视为是统计能量的可能公式的全部。例如,可选地,可以采用如下公式来定义统计能量:
[0122][0123]
例如,在多重比对(参见上面的定义)中没有对序列进行比对时可以使用该公式。
[0124]
除了依赖于序列的参数θ之外,还有项uk,其表示取决于序列长度l的能量统计贡献。
[0125]
还可能的是,例如当序列不是特别长时,只采用独立位置偏好(即,只是根据上述第一个实施例的表达式的第一个项)来定义统计能量。
[0126]
通常,在分子的令人感兴趣的至少一个理化特性的筛选实验过程中,针对预定选择来计算参数。
[0127]
t是循环(round)/循环(cycle)的数目
[0128]
c是靶标的数目
[0129]
s(n(t),c)是如下定义的函数:如果那么s(n(t),c)=1,其它情况下,s(n(t),c)=0。
[0130]
参考筛选实验,将概率定义为选择和扩增的t个循环的联合概率,如下(等式1):
[0131][0132]
其中,p
reg
(p)是正规项,项p(n(0))是指零循环中存在的载体的分布。其它三个因子具有如下定义:
[0133]
读取因子p(r(t)
│
n(t))是从载体的分布n={ns(t)}提取一组读数r={rs(t)}的概率,由如下方程定义:
[0134][0135]
其中,r
tot
(t)是循环t中读取的总数
[0136]
第二项是扩增因子,即从循环t中选择的n(t)个载体开始在循环t+1中扩增n(t+1)个载体的概率,定义如下:
[0137][0138]
第三项是指选择,表示从n(t)个存在的载体中选择n(t)载体的概率,定义如下:
[0139]
[0140]
学习或训练程序包括:通过已知的优化算法查找使后验联合概率p最大化的能量参数的标量值,以及指定从筛选实验读取r={rs(t)}训练实验数据。
[0141]
一种优选实施方案的实施例:具有罕见且确定的联系的两状态系统,对两点相互作用的上位性效应的描述
[0142]
在一种优选的实施方案中,有可能只考虑两种状态,即,选择的和未选择的(例如,与靶标相连或者未与靶标相连)。
[0143]
此外,假定结合分子的情况是罕见现象,因此,概率远小于1。
[0144]
进一步的假定是无穷多的靶标c
→
∞,如果靶标数目远高于筛选实验中存在的载体的数目,那么该近似值是现实可行的。这种情况在大部分实验中得到证实。
[0145]
在这种情况下,去除状态指数k,因为存在k-1个统计能量存在,并且,在这种情况下,状态是2。
[0146]
将ns(t)作为循环t中与序列s结合的载体的数目。
[0147]
根据这样的假定,将上面描述的三个因子简化为如下:
[0148]
考虑到r(t)≈n(t),去除读取因子p(r(t)
│
n(t))。
[0149]
扩增因子变成:
[0150][0151]
选择因子变成:
[0152][0153]
其中,ps是选择序列s的概率,将其定义为其中es=e
结合
(s)-e
未结合
(s)。
[0154]
考虑到ps《《1,可以近似为
[0155]
在当前的实施例中,用一个和两个位点的相互作用使能量参数化:
[0156][0157]
该表达式连同选择序列的概率共同构建了基因型-表型图谱,原因是它将序列(基因型)与处于一种分子状态(表型)的概率联系在一起。由此,联合概率的对数变为:
[0158][0159]
采取的近似值使的最大值弱化了参数θi和θ
ij
的优化问题。一旦通过实验选择的序列已经被指定为n(t),那么使用l-bfgs优化算法能够解决前述问题。
[0160]
也可以说,即使是最普遍的问题,即试图通过以更一般的形式(即等式1)使似然函数最大化从而计算参数θi和θ
ij
,也能够通过数值方法得以解决。
[0161]
用于训练的输入数据
[0162]
输入数据来自于生物聚合物分子的变体的筛选实验,例如,深度突变扫描、定向进化或基于selex的技术。
[0163]
输入数据是生物序列(例如实验中使用的突变体的氨基酸或核苷酸)以及选择循环的计数。
[0164]
这些可以从测序数据获得(例如,fastq格式的dna读取(reads))。
[0165]
如图2描述,典型的生物信息学程序对应于在四个数据集中进行的预处理,这四个数据集用作下表1概括的测试。
[0166]
测序由dna丝状结构(dna filaments)构成,从每个循环的读数集开始,例如采用fastq格式,该程序具有如下步骤:
[0167]
·
对低测序质量的读取或者正向和反向读取不匹配的读取进行过滤;
[0168]
·
将核苷酸序列翻译成氨基酸序列,去除有终止密码子的序列;
[0169]
·
计算每个循环中序列的数目;
[0170]
·
对各种循环中出现的总数小于10的序列进行过滤。
[0171]
概率模型的训练
[0172]
如前几段所述,该步骤对使似然函数最大化的问题进行了数值求解,将其求解为上述选择因子的至少一个统计能量函数的参数函数。因此得到的参数最佳值具有进入模型的训练序列集的特征,如果对这些训练序列进行修饰,参数最佳值会改变。
[0173]
统计能量函数的参数的用途
[0174]
因此,上述的概率统计方法对来源于突变体生物序列的选择与富集实验的测序数据文库进行分析,目的是评价至少一个输入的突变体生物序列并且针对指定的理化特性选择最佳序列。通过允许的分子状态,采用统计能量函数或统计能量的组合对该特性进行量化,其中,通过在学习阶段计算每个统计能量函数的能量参数来定义前述的统计能量的组合。
[0175]
随后,从这些参数开始,可以生成具有令人感兴趣的理化特性的生物序列文库。
[0176]
然后,模型适用于:
[0177]-基于给定的特性来评价突变体并选择最佳突变体。尤其,一旦已经确定了上述定义的能量参数,就可以将这些参数用来计算与输入序列有关的至少一个统计能量函数,作为新序列的非限制性实例,即不属于训练步骤中使用的序列。一旦通过使似然函数最大化计算了相关能量参数,统计能量实际上是未知的生物序列的函数。能量函数的每一个都与选择实验过程中观察到的相关分子状态有关,由这些能量函数,可以得到分子状态所指的与所期望理化特性相关的序列的分数;
[0178]-产生生物序列文库,例如具有给定理化特性的蛋白质的生物序列文库,推断一组具有统计能量优化函数特征的序列。
[0179]
突变体评价
[0180]
在本发明方法的一种优选实施方案中,根据相关编码蛋白质的特征或活性计算给定生物序列(氨基酸或核苷酸)的得分。待评价的生物序列可来自于训练数据(图2右侧方框的第一行)或来自于其它实验,这些实验的数据尚未用于学习模型(图2右侧方框的第二行)。
[0181]
在上述构建的两种状态结构中(例如,存在联系或无联系),得分例如由统计能量
本身来确定:
[0182][0183]
在三种状态(即,结合的、非结合的和非折叠的)的一种实施方案中,可将得分φ定义为结合状态e
l
和折叠状态ef的能量的如下组合:
[0184][0185]
只通过选择与输入突变体序列有关的统计能量参数并应用得分方程式,就能计算得分,在两种状态实施例的情况中,得分方程式对应于上述段落中统计能量函数的表达式。
[0186]
尤其,前述三种状态得分的分值高相当于给定实验中相关分子处于结合且折叠状态的概率高。另一方面,如果将得分定义为与统计能量相同,那么低分值表示处于结合状态的概率高。
[0187]
一个或多个最佳序列的产生
[0188]
在本发明方法的一个优选实施方案中,有可能产生使模型的得分函数φs最大化的序列。通常,从每个考虑到的状态k的能量函数ek(s)开始定义得分函数φs。
[0189]
通过序列的搜索算法来产生具有最佳得分的序列,该搜索算法以绝对或相对的方式使指定的得分函数φs最大化。其有效算法是模拟退火,一种标准的优化算法。
[0190]
根据一种优选的实施方案,数据来源于dms实验,该dms实验使用了(fowler,douglas m.,and stanley fields."deep mutational scanning:a new style of protein science."nature methods 11.8(2014):801.)中描述的蛋白质展示技术中的一种。
[0191]
根据另一种优选的实施方案,数据来源于dms和定向进化(de)实验,这些实验的目的是选择能结合特定分子靶标的有效蛋白质变体,优选肽或蛋白质,该方法的目的是在被分析的变体中选出最具选择性的蛋白质变体。
[0192]
根据另一种优选的实施方案,数据来源于dms和de实验,这些实验的目的是选择能有效结合特定分子靶标的蛋白质变体,优选肽或蛋白质,该方法的目的是产生针对分子靶标具有更高选择性的蛋白质变体文库。
[0193]
根据另一种优选的实施方案,数据来源于dms和de实验,这些实验的目的是选择能有效进行特定催化作用的蛋白质变体,根据本发明的方法的目的是产生在所述酶催化作用中具有更高活性的蛋白质变体文库。
[0194]
根据另一种优选的实施方案,数据来源于dms和de实验,这些实验的目的是选择能有效进行特定催化作用的蛋白质变体,根据本发明的方法的目的是从文库中选出在酶催化作用中活性最高的蛋白质变体。
[0195]
根据另一种优选的实施方案,数据来源于dms和de实验,这些实验的目的是选择能有效实现最佳光致发光的蛋白质变体,根据本发明的方法的目的是从文库中选出最具光致发光的变体。
[0196]
根据另一种优选的实施方案,数据来源于dms和de实验,这些实验的目的是选择在高温条件下具有活性的蛋白质变体,根据本发明的方法的目的是从文库中选出耐热性最强
的变体。
[0197]
根据一种优选的实施方案,数据来源于本领域技术人员已知的selex实验或基于其的技术;作为非限制性实例,参见“zhuo z,et al.-recent advances in selex technology and aptamer applications in biomedicine int j mol sci.2017oct 14;18(10)”。
[0198]
根据另一种优选的实施方案,数据来源于selex实验或基于其的技术,这些实验或技术的目的是选择能有效结合特定分子靶标的适体,该方法的目的是在被分析的适体中选出最具选择性的适体。
[0199]
根据另一种优选的实施方案,数据来源于selex实验或基于其的技术,这些实验或技术的目的是选择能有效结合特定分子靶标的适体,该方法的目的是产生针对分子靶标具有更高选择性的适体变体文库。
[0200]
根据另一种优选的实施方案,将该方法用于所谓的“机器学习辅助定向进化”过程。因此,在这种实施方案中,根据称作机器学习辅助定向进化的方案,该方法采用来自一个或多个定向进化循环的数据进行训练,并将该方法用于产生有效的蛋白质变体,以便在随后的定向进化循环中进行改变和测试。
[0201]
因此,对于蛋白质或核苷酸序列的文库的计算机模拟筛选而言以及对于选择具有期望特性的突变体而言,本发明的方法是有效且可靠的,其中,前述文库是从dms、de实验或从基于selex的技术获得的。根据本发明的方法,还可能将来自这些实验的数据用于得到分子理化特性的高效率序列的文库,其中高效率的意思例如是高催化能力、高适应度、与特定分子靶标结合的能力高。
[0202]
通常,根据本发明的方法可用于所有类型的dms或de实验,这些实验至少具有一个选择循环。
[0203]
通常,根据本发明的方法可用于所有类型的hts-selex(高通量测序selex)实验和由其衍生的技术,这些实验和技术至少具有一个选择循环。
[0204]
实施例
[0205]
下面的实施例是对本发明的举例说明,不应认为是对相关保护范围的限制。
[0206]
下面,我们使用来源于四个dms实验的数据报告了该方法的性能,dms实验简要描述如下,其特征总结如下表所示。
[0207]
表i
[0208][0209]
实施例1.利用噬菌体展示法进行的dms实验数据预测突变抗体结合的选择性
[0210]
该模型已经在s.boyer et al.“hierarchy and extremes in selections from pools of randomized proteins.”pnas(2016)公布的数据上进行了测试。
[0211]
报导的dms实验的目的是分析抗体文库并与中性的合成聚合物聚乙烯吡咯烷酮(pvp)结合。在这种情况下,使用噬菌体展示技术进行三个循环的扩增和选择来实施实验。通过在决定互补性3(cdr3)的区域的四个连续氨基酸上饱和诱变抗-pvp抗体来创建初始文库。
[0212]
实施例2.hyap65蛋白质ww结构域突变体的结合选择性的预测,数据来源于使用噬菌体展示法进行的dms实验
[0213]
该模型已经在d.m.fowler et al.“highresolution mapping of protein sequence-function relationships.”nature methods(2010)公布的数据上进行了测试。
[0214]
报导的dms实验的目的是对选择用于与肽配体(gtppppytvg)结合的ww结构域突变体的文库进行分析。使用噬菌体展示技术进行6个循环的扩增和选择并且进行3轮测序(0.3,6),从而实施实验。采用“掺杂寡核苷酸合成”技术来创建初始文库。
[0215]
实施例3.hyap65蛋白质ww结构域突变体的结合选择性的预测,数据来源于使用噬菌体展示法进行的dms实验
[0216]
该模型已经在c.l.araya et al.“a fundamental protein property,thermodynamic stability,revealed solely from large-scale measurements of protein function.”pnas(2012)公布的数据上进行了测试。
[0217]
报导的dms实验的目的是对选择用于与肽配体结合的ww结构域突变体的文库进行分析。在这种情况下,使用噬菌体展示技术进行4个循环的扩增和选择,从而实施实验。通过采用“掺杂核苷酸库”(doped nucleotide pools)化学合成dna来创建初始文库。
[0218]
实施例4.结合结构域的突变体与g蛋白的免疫球蛋白g(igg)(蛋白g的igg-结合结构域)(gb1)的结合选择性预测,数据来源于mrna展示法进行的dms实验
[0219]
该模型已经在c.olson et al.“a comprehensive biophysical description of pairwise epistasis throughout an entire protein domain.”current biology(2014)公布的数据上进行了测试。
[0220]
报导的dms实验的目的是对与igg-fc结合中选择的gb1蛋白突变体文库进行分析。在这种情况下,使用mrna展示进行扩增和选择循环,从而实施实验。采用饱和诱变技术创建初始文库。
[0221]
测试类型
[0222]
将来源于这些实验的数据集随机分为模型的训练集和测试集,其中,将模型的统计结合能与突变体结合能力的实验测量值进行比较,然后选择突变体进行下一个循环。将突变体选择性的这种测量定义为平均连续两个循环中序列在一个循环出现频率与下一个的比值。在公式中,我们将序列的选择性s定义为:
[0223][0224]
其中,fs(t)是第t个循环中检测的序列s的频率,t是循环的总数(图4、5和6)。
[0225]
该方法的实施方案在上述段落中进行了报导,这些段落涉及具有稀有确定性键的两状态系统和对双位点相互作用的上位性效应的描述。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1