胰液素微粒的制作方法
1.本发明涉及医药领域,并且涉及为包有肠溶包衣的微粒的口服剂型的基于胰液素(pancreatin)的酶制品。
2.最具体而言,本发明描述了一种药物组合物,其被制造为包括微粒芯部,该药物组合物具有药学有效量的胰液素、十六烷醇和泊洛沙姆407。
3.本发明涉及所述药物组合物的制造方法,以及基于药物组合物的用水基肠溶包衣包覆的微粒的制造。
4.通过根据本发明的方法获得包有肠溶包衣的微粒,并且该肠溶包衣微粒在儿童和成人的替代疗法中用作治疗由于胰酶活性下降而引起的外分泌(酶)胰腺功能不全的药品。胰酶活性下降是由于生产、分泌调节和胰酶递送的紊乱、或由于胰酶在肠道中的破坏增加而导致的,其中胰酶在肠道中的破坏增加由胃肠道的各种疾病引起,最常见于胰腺手术后、胃切除术后的囊性纤维化、慢性胰腺炎;胰腺癌;胃的部分切除(例如,billroth ii)。
背景技术:
5.已知胰液素是各种具有生理活性的消化酶如脂肪酶、淀粉酶和蛋白酶的混合物。胰脂肪酶、淀粉酶和蛋白酶是治疗诸如胰腺外分泌功能不全的不同病理状况的活性消化酶补充剂。
6.消化酶为胰酶,并且是指存在于胰腺分泌物中的任何类型的酶,例如淀粉酶、脂肪酶、蛋白酶、或它们的混合物、或者任何具有酶活性的胰腺提取物,例如胰液素。适合于本发明的应用的消化酶的种类可以至少包括脂肪酶、淀粉酶和蛋白酶。
7.胰液素或消化酶通常被制成片剂、胶囊剂或颗粒剂的形式,其必须满足多种药品要求,其中存在以下要求,根据这些要求,片剂、胶囊剂或颗粒剂应当保护内部所含的酶在通过ph=1.0的胃时以及在ph=5.0或ph=6的上肠时不降解,并且当其在进入ph=6.0的小肠时溶解,从而以所需量释放活性酶并且用于患者是安全的、特别是无毒的。
8.在摄入过程中酶活性的维持以及在肠中的快速释放、特别是稳定性和溶解性是制品的重要特征。在许多研究中,发现例如颗粒、微粒或小丸是将酶递送至肠的最有效形式。
9.有许多已知的用于制造颗粒或小丸的方法,其包括以下步骤:制备可挤出的混合物,挤出小丸的芯部,干燥所得的小丸的芯部,施加肠溶包衣,以及干燥小丸。
10.例如,欧洲专利ep 0583726描述了具有65重量%至85重量%、特别是具有75重量%至80重量%的胰液素的包有肠溶包衣的胰液素小丸,该包有肠溶包衣的胰液素小丸的体积密度为0.6g/ml至0.85g/ml,并且主要包含胰液素、聚乙二醇4000和低粘度石蜡,相对于每100重量份的胰液素,包含:15重量份至50重量份(特别是20重量份至30重量份)的聚乙二醇4000和1.5重量份至5重量份(特别是2重量份至3重量份)的低粘度石蜡,其特征在于呈球形或椭球形,同时球体的直径或椭球的短轴为0.7mm至1.4mm、优选0.8mm至1.2mm,并且其特征在于,其中至少80%的胰液素微丸的粒度分布的特征在于椭球的短轴与椭球的长轴的比例范围为1:1至1:2。
11.这些酶制品的缺点在于,在成分组成中存在低粘度石蜡和矿物油、特别是凡士林,这至关重要,因为目前不推荐为孕妇或婴儿开矿物油处方。
12.专利ru 2440101c2描述了一种包含胰液素与肠溶包衣的口服剂型的药物组合物,其中肠溶包衣包含成膜剂、十六烷醇和作为增塑剂的乙酸三乙酯,以及至少一种抗粘剂。
13.最接近的现有技术是专利ru 2408364 c2,其描述了一种用于制造胰液素微丸的芯部的方法及其应用。此外,该专利描述了由成膜剂和增塑剂组成的胰液素微丸的肠溶包衣。
14.以已知的方式,可以获得胰液素微丸的芯部,其包含70重量%至90重量%的胰液素、10重量%至30重量%的至少一种药学上可接受的粘合剂、以及至多5重量%的至少一种药学上可接受的惰性填充剂。
15.该制造方法的缺点在于,在所得的微丸中存在痕量的丙酮,并且在ph=1至6的条件下酶缺乏稳定性。
16.在根据上述胰液素微丸的方法获得的组合物中出现残留的丙酮使得该制品对患者不安全,因为丙酮具有第3级别的毒性,并且可以引起一些器官(例如,皮肤和肺)的损伤。尽管事实上在此类微丸的组合物中残留的丙酮的含量很少,但是在治疗许多疾病、特别是囊性纤维化时,长期或甚至终生应用多剂量(在一天中)的此类微丸会导致其他健康威胁。
17.因此,本发明的目的是提供一种新的药物组合物,以用于制造口服剂型微粒的具有胰液素的酶制品,该微粒具有低毒性、即不存在残留量的丙酮,具有高稳定性和溶解性,由于具有肠溶包衣的口服剂型中的成分的定性和定量组成,因此该微粒的应用对所有年龄的患者和来自所有特定群体的患者都没有限制。
18.所述发明的一个实施方案是具有肠溶包衣的胰液素微粒的口服剂型。
19.由于包含预定量的胰液素、十六烷醇、泊洛沙姆407的微粒芯部的新组合物、用于制造这种芯部的新方法、以及利用肠溶水基包衣制造微粒本身,这个问题得以解决。此外,所得的口服剂型不含残留量的丙酮和合成油。
技术实现要素:
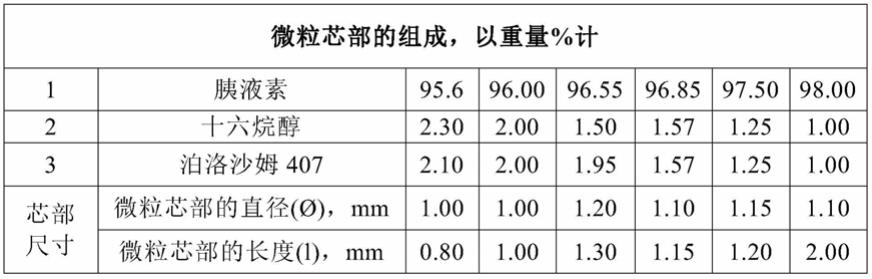
20.在本发明更具体的优选实施方案中,提供了用于制备治疗与胰腺外分泌功能不全、消化不良、胰腺炎、囊性纤维化、i型糖尿病和/或ii型糖尿病相关的消化系统紊乱的药品的口服剂型(微粒芯部),其包含95.6重量%至98.0重量%的胰液素、1.0重量%至2.3重量%的十六烷醇和1.0重量%至2.10重量%的泊洛沙姆407。因此,在球化阶段之后获得的芯部具有以下尺寸:d(芯部直径)=1.0mm至1.2mm,l(芯部长度)=0.8mm至2.0mm。
21.泊洛沙姆407是包含两个亲水性聚乙二醇(peg)嵌段以及位于它们之间的中央疏水性聚丙二醇(ppg)嵌段的三嵌段共聚物。
22.此外,本发明涉及一种由形成微粒芯部所必需的粘合剂制造药物组合物的方法。首先,特别地,制备粘合剂混合物,该粘合剂由乙醇、十六烷醇、泊洛沙姆407这三种组分以1:0.11:0.11至1:0.13:0.13的比率范围组成,对粘合剂混合物施加一定的技术条件,例如同时保持工艺温度在40℃至45℃的范围内。在溶剂、特别是乙醇的存在下,以预定的最佳量将胰液素与所得的粘合剂混合物混合。此外,在该方法的优选实施方案中,以严格的顺序进行药物组合物的制造阶段,其包括将乙醇添加到有效量的胰液素中,然后将预先形成的三
组分粘合剂引入体系中。
23.在获得均匀的混合物后,在乙醇的存在下使微粒芯部成形并球化。此外,对微粒芯部进行干燥,在此期间除去乙醇,并将温度保持在在34℃。通过将肠溶包衣溶液施加至芯部,从而获得胰液素微粒。
24.在本发明的一个实施方案中,肠溶包衣包含水、聚乙二醇4000、滑石、二甲基硅油的乳液、比例为1:1的甲基丙烯酸和丙烯酸乙酯共聚物的悬浮液。该药物组合物和/或微粒以适合于口服给药的剂型使用,并可以制备用来治疗与胰腺外分泌功能不全、消化不良、胰腺炎、囊性纤维化、i型糖尿病和/或ii型糖尿病相关的消化系统紊乱的药品。
25.该药物组合物和/或微粒以适合于口服给药的剂型使用,并可以制备用来治疗与胰腺外分泌功能不全、消化不良、胰腺炎、囊性纤维化、i型糖尿病和/或ii型糖尿病相关的消化系统紊乱的药剂。此外,将胰液素与乙醇以严格的顺序混合,根据该顺序,首先取胰液素,然后添加乙醇,之后将三组分粘合剂引入体系中,使得所得的悬浮液具有均一性。发现在悬浮液的制备过程中逐渐(不连续地)引入粘合剂使得悬浮液高度均匀。
26.此外,十六烷醇(脂肪醇)作为粘合剂的这一功能性用途促进了微粒芯部形成过程中的压实并提高了颗粒之间的粘附力。
27.添加泊洛沙姆407,其作为粘合剂和增溶剂,强化了胰液素微粒芯部,并增加了活性药物成分、即胰液素的溶解。
28.下面提供的研究结果表明,本发明能够制备这样的胰液素微粒,其中该胰液素微粒在ph=1.0时对胃酸的作用具有高耐受性,并且在ph=6.0时具有相当高的溶解速率(不超过30分钟)。
29.将肠溶包衣施加至具有胰液素的药品的口服剂型,必须将该胰液素递送到胃肠道中ph大于胃中ph的区域。本发明所要求保护的肠溶包衣的组成是水基的,进而在ph=6.0时提供了良好的溶解性,而不损失微粒芯部的酶活性,并且(例如)与存在丙酮残留物的酶制品相比无毒性。
30.根据本发明,出乎意料的是发现根据所要求保护的方法制备的胰液素微粒芯部适合施加肠溶包衣,并且具有高稳定性,同时保持了酶活性。还发现,就所使用的组分的安全性而言,与其他已知方法相比,本发明所述的制备微粒芯部的方法更有效。特别地,与其他已知的肠溶包衣的组成不同,所建议的胰液素微粒芯部的肠溶包衣不含有机溶剂,例如具有毒性的丙酮。出乎意料的是,发现与基于丙酮的肠溶包衣的组成相比,基于水的肠溶包衣的组成在其性质上并不差。此外,将肠溶水基包衣施加至胰液素微粒对制品稳定性有积极影响。胰液素微粒芯部的储存稳定性示出了良好的结果,这是由于在芯部的组合物中使用了预定比例的十六烷醇和泊洛沙姆407。
31.根据本发明,出乎意料的是发现作为有肠溶包衣的微粒的芯部的一部分的药物组合物在胃酸(ph=1.0)以及上肠(ph=6.0)中是稳定的,上述药物组合物包含水、聚乙二醇4000、滑石、二甲基硅油的乳液、甲基丙烯酸和丙烯酸乙酯的悬浮液。
32.在本发明的其他实施方案中,微粒的肠溶包衣可以是水基的,特别是包含药学有效量的水、柠檬酸三乙酯、滑石、二甲基硅油、甲基丙烯酸和丙烯酸乙酯的混合物。
33.为了获得口服剂型,应当形成药剂制品微粒并将所得的混合物与乙醇一起球化,在该过程后,应当在完全除去所用乙醇的条件下进行芯部干燥。成形为挤出所得的胰液素
和粘合剂的混合物的过程。根据本发明,形成的胰液素芯部的最佳干燥温度保持在约34℃。该温度方案确保了更好地保存酶组分。
34.此外,本发明涉及用于制造具有肠溶包衣的胰液素微粒的方法,该肠溶包衣包含药学上可接受的量的水、乙酸三乙酯、滑石、二甲基硅油、甲基丙烯酸和丙烯酸乙酯的混合物。微珠也不包含残留丙酮。
35.本发明能够获得这样的胰液素微粒,其在ph=1.0时对胃酸作用具有高耐受性,并且在ph 6.0时在不超过30分钟时快速溶解。
36.出乎意料的是,发现与基于丙酮的肠溶包衣的组成相比,基于水的肠溶包衣的组成在其性质上并不差。出乎意料的是,发现与基于丙酮的肠溶包衣的组成相比,基于水的肠溶包衣的组成在其性质上并不差。此外,将肠溶水基包衣施加至胰液素微粒对制品稳定性有积极影响。胰液素微粒芯部的储存稳定性示出了良好的结果。
37.如本发明所述,当在微粒上施加肠溶包衣时,水用作主要溶剂。这可以通过以下原因来解释,分别为:没有暴露于危险因素的风险,例如蒸汽的毒性作用、爆炸和火灾;非防爆设备和较廉价的防火场所;不需要研究制品中残留溶剂的存在且无毒性。
38.因此,在本发明中,获得了微粒芯部的主要组分的最佳比例和引入粘合剂的特定顺序,这产生了能够使消化酶型胰液素处于稳定状态、在典型的储存条件下提供在肠道内的有效释放且活性损失最小的药物组合物。
39.本发明的技术效果是分别获得了芯部和肠溶包衣微粒的较高稳定性,同时保持了具有肠溶包衣的微粒的良好溶解性,这使得可以应用所要求保护的胰液素微粒以制备安全且无毒的药品,用来治疗由胰腺外分泌功能不全、消化不良、胰腺炎、i型和/或ii型糖尿病引起的消化系统紊乱。用于制造芯部和具有水基肠溶包衣的微粒的方法的技术条件使得可以实现药品的高溶解性、储存稳定性而不损失酶活性,并且确保对于需要治疗消化系统紊乱的所有年龄群体的患者而言均为安全应用。
40.通过该方法获得的胰液素微粒的测试表明,与其他已知的使用其他粘合剂的胰液素微丸相比,胰液素微粒保持了更高的脂肪酶含量。
41.因此,本发明所述的胰液素微粒对胃酸具有高耐受性,并且(例如)在ph=1和/或ph=6时具有保护能力。
42.根据本发明,胰液素微粒的干燥温度保持在35℃至50℃的范围内以确保更好地保存酶组分。
具体实施方式
43.实施例1(根据本发明)
44.实施例说明了在乙醇溶剂的存在下制备包含胰液素的药物组合物,其中可获得微粒芯部。
45.首先,制备粘合剂的溶液。
46.根据本发明的一个实施方案,在40℃至45℃将乙醇注入配备有搅拌器和加热夹套的制造容器中。然后在搅拌下,以乙醇:十六烷醇:泊洛沙姆407为1:0.11:0.11的比例分别添加十六烷醇(粉末)、泊洛沙姆407(粉末)。由此获得粘合剂三元混合物。
47.根据本发明的一个实施方案,在40℃至45℃将乙醇注入配备有搅拌器和加热夹套
的制造容器中。然后在搅拌下,以乙醇:十六烷醇:泊洛沙姆407为1:0.12:0.12的比例分别添加十六烷醇(粉末)和泊洛沙姆407(粉末)。
48.根据本发明的其他实施方案,在40℃至45℃将乙醇注入配备有搅拌器和加热夹套的制造容器中。接下来,在搅拌下,以乙醇:十六烷醇:泊洛沙姆407为1:0.13:0.13的比例分别添加十六烷醇(粉末)和泊洛沙姆407(粉末)。
49.根据本发明的一个实施方案,将胰液素装入混合制粒机中,然后添加乙醇以改善润湿性,然后将制备好的粘合剂在环境温度下装入考虑了上述比例的润湿混合物中。在每次供应一种组分后,彻底搅拌混合物。所限定的一致供应的组分、特别是将胰液素与乙醇预混合,确保了组分的均匀溶解和微粒芯部中的组分更有效的相互作用。
50.使用具有1.0mm孔径的模具,并且在颗粒的控制温度为30℃的情况下,将所得的混合物装入以进行挤出。将直径为1.0mm至1.2mm且长度为0.8mm至2.0mm的所得的芯部供应至球化步骤,球化步骤在浓度为55重量%至96重量%的乙醇溶剂的存在下进行,其中胰液素与乙醇的比例为1:0.38。
51.在实施该方法的过程中,获得了微粒芯部,其组成示于下表。
52.表1
[0053][0054]
球化后,在温度为34℃、湿度为2重量%至5重量%时对所得的微粒芯部进行干燥。供应经干燥的芯部,用于接下来的肠溶包衣溶液的施加步骤。
[0055]
由下列成分混合从而制备肠溶包衣溶液:比率为1:0.03:0.14:0.004:1.04的水、聚乙二醇4000、滑石、30%二甲基硅油的乳液、甲基丙烯酸和丙烯酸乙酯共聚物(1:1)的悬浮液(30%分散体)。通过在微粒与所得的溶液的比例为1:1.57的情况下,对预热至35℃的颗粒进行喷涂,从而将制备好的溶液施加至微粒芯部上。
[0056]
在施加肠溶包衣溶液的步骤结束时,将所得的微粒干燥,同时保持温度在35℃至50℃的范围内。
[0057]
实施例2(通过制造微粒芯部的方法进行比较)
[0058]
用于制造胰液素微粒芯部的方法与实施例1类似,不同之处在于,首先,将乙醇装入混合制粒机中,并且分批添加胰液素,然后以实施例1所述的比例添加粘合剂。在每次供应一种组分后,将混合物彻底混合15分钟。
[0059]
实施例3(通过制造微粒芯部的方法进行比较)
[0060]
用于制造胰液素微粒芯部的方法与实施例1类似,不同之处在于,将制备好的胰液素与粘合剂的混合物装入成形机的料斗中。使用孔径为1.0mm的模具,在控制芯部的干燥温度为28℃的情况下,将悬浮液制粒。
[0061]
实施例4(通过制造微粒芯部的方法进行比较)
[0062]
用于制造胰液素微粒芯部的方法与实施例1类似,不同之处在于,将制备好的胰液素与粘合剂的混合物装入成形机的料斗中。使用孔径为1.0mm的模具,在控制芯部的干燥温度为41℃的情况下,将悬浮液制粒。
[0063]
实施例5(通过制造微粒芯部的方法进行比较)
[0064]
用于制造胰液素微粒芯部的方法与实施例1类似,不同之处在于,将制备好的胰液素与粘合剂的混合物装入成形机的料斗中。使用孔径为1.0mm的模具,在控制芯部的干燥温度为38℃的情况下,将悬浮液制粒。
[0065]
实施例6(根据本发明)
[0066]
根据实施例1所述的制造胰液素微粒的方法,不同之处在于肠溶包衣溶液中的成分比例,水:聚乙二醇4000:滑石:30%二甲基硅油的乳液:甲基丙烯酸和丙烯酸乙酯共聚物(1:1)的悬浮液(30%分散体)的比率为1:0.03:0.16:0.004:1.03。将所得的溶液搅拌15分钟。
[0067]
实施例7(与原型比较)
[0068]
根据原型所述的制造微丸的方法,其中将丙酮用作肠溶包衣的制备中的溶剂。
[0069]
1.胰液素微丸的制备:
[0070]
在混合器中将1.59kg的胰液素与0.25kg的聚乙二醇4000混合,用0.25kg的2-丙醇充分润湿。将所得的混合物成形为内径为1.0mm。在压制过程中,温度低于50℃。将成形后所得的1.46kg的胰液素以等份送至球化步骤从而获得微丸芯部。在球化过程中,添加约13.5g的2-丙醇。在温度范围为35℃至50℃干燥12小时后,通过在筛上筛分而分选胰液素微粒。
[0071]
2.肠溶包衣的施加:
[0072]
通过在室温搅拌下添加231.4g的邻苯二甲酸羟丙基甲基纤维素、12.85g的柠檬酸三乙酯、4.89g的十六烷醇和5.55g的二甲基硅氧烷、1000g至2000g的丙酮,从而制备包衣溶液。通过将所得的溶液喷涂在胰液素微丸上,施加包衣直至成膜。在包衣过程中微丸芯部的温度保持在37℃至43℃的范围内。然后将所得的微丸在温度范围为35℃至50℃干燥12小时。
[0073]
根据本发明获得的胰液素微粒的稳定性测试
[0074]
对于通过实施例1至7所述的方法获得的胰液素微粒芯部,在模拟ph=1.0的胃环境和ph=6.0的上肠环境的条件下进行稳定性测试。
[0075]
通过生物化学方法,根据sp xiii gpm 1.4.2.0014.15“固体药物剂型的溶出度(dissolution for solidpharmaceutical dose forms)”,在ph=1.0和ph=6.0时进行活性物质的量的确定,该活性物质在一定时间段内必须从固体剂型中释放到溶出介质中。
[0076]
溶出度试验分两步进行。
[0077]
步骤1(酸性,即ph=1.0),使用叶轮混合器。
[0078]
4m氢氧化钠溶液。在37
±
0.5℃的温度范围内,将16.0g的氢氧化钠置于100ml容量瓶中,溶解在80ml的水中。将溶液冷却至室温后,将溶液稀释至所需体积并混合。测试时间为120分钟。
[0079]
步骤2(碱性,即ph=6.0),使用叶轮混合器。
[0080]
ph=6.0的磷酸盐缓冲液,其中将2.0g的氯化钠和9.2g的磷酸二氢钾置于1000ml
容量瓶中并溶解在约950ml的水中。在37
±
0.5℃的温度,使用4m氢氧化钠溶液电位滴定调节溶液的ph至6.0,将溶液稀释至所需体积。溶解过程持续30分钟。
[0081]
在ph=1.0持续120分钟和在ph=6.0持续30分钟测试样品的过程中获得的平均值示于表2至表4。
[0082]
以在孵育后残留的脂解活性相对于孵育前测试的样品的实际脂解活性的百分比给出所制备的样品在模拟胃环境的条件下的溶出稳定性特征。
[0083]
根据上述gpm“固体药物剂型的溶出”,在胃中释放时释放到溶出介质中的活性物质的量不应当超过所要求保护的胰液素含量的10%;在上肠环境中释放时释放到溶出介质中的活性物质的量应当为所要求保护的胰液素含量的至少75%。
[0084]
通过生物化学方法确定脂解活性,并与标准样品的胰液素的酶的比活性相比较。通过比较胰液素微粒悬浮液水解橄榄油乳液底物的速率与标准胰液素(脂肪酶)样品的悬浮液在相同条件下水解相同底物的速率,从而确定脂解活性。
[0085]
在模拟胃环境和上肠环境的条件下,比较根据实施例1和2的胰液素微粒的溶出稳定性指标。
[0086]
表2
[0087][0088]
溶出度试验的结果表明,根据实施例1的组合物在ph=1.0和ph=6.0的耐胃酸性超过实施例2中获得的组合物的稳定性指数。样品在ph=1的稳定性应当不超过10%,这是对照选择。
[0089]
根据《国家药典》,xiii,ofs,1.4.2.0014.15“固体剂型的溶出”的药典通则,当在胃中释放时,释放到溶出介质中的活性物质的量应当不超过所要求保护的胰液素含量的10%;在上肠环境中释放到溶出介质中的活性物质的量必须是所要求保护的胰液素含量的至少75%。
[0090]
在模拟胃环境和上肠环境的条件下,比较根据实施例3、4和5的胰液素微粒的溶出稳定性指标。
[0091]
表3
[0092][0093]
当比较不同的干燥温度时,根据实施例3、4和5制备的组合物在模拟胃和上肠环境的条件下获得的胰液素微粒溶出稳定性指标的比较结果清楚地示出了在球化后选择34℃的温度干燥微粒芯部的优势。
[0094]
比较根据实施例1和2制备的组合物和原型在ph=1.0和ph=6.0时的耐胃酸性试验结果。
[0095]
表4
[0096][0097]
比较胰液素微粒在模拟胃环境和上肠环境的条件下溶出时的稳定性指标的结果,与原型组合物相比,根据实施例1和2制备的组合物示出了所提出的制造微粒芯部的药物组合物的方法的优势。
[0098]
因此,发现胰液素微粒的测试样品在ph=1.0时耐胃酸,并且根据胰液素标准品的预定脂解活性,胰液素微粒的测试样品的活性不低于:
[0099]
对于根据实施例1制备的样品为4.6%(甚至更优选的值);
[0100]
对于根据实施例4制备的样品为7.0%(优选值);
[0101]
对于根据实施例2制备的样品为6.8%(优选值);
[0102]
对于根据实施例5制备的样品为6.4%(更优选值);
[0103]
对于根据实施例3制备的样品为6.1%(更优选值)。
[0104]
本发明的胰液素微粒在ph=6.0的上肠中释放时是稳定的,并且根据胰液素标准品的预定脂解活性,本发明的胰液素微粒的效率不低于:
[0105]
对于根据实施例1制备的样品为93.7%(甚至更优选的值);
[0106]
对于根据实施例5制备的样品为89.5%(更优选值);
[0107]
对于根据实施例3制备的样品为87.3%(更优选值);
[0108]
对于根据实施例4制备的样品为84.6%(优选值);
[0109]
对于根据实施例2制备的样品为83.8%(优选值)。
[0110]
胰液素微粒芯部的储存稳定性示出了良好的结果。
[0111]
出乎意料的是,发现与丙酮基包衣相比,肠溶水基包衣在其性质上并不差。随后,在水性溶剂的存在下将肠溶包衣施加在胰液素微粒上对药物组合物在控制点的储存稳定性产生了积极影响。
[0112]
表5
[0113]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1