通过阿片类己二烯酸酯和任选取代的己二烯酸酯增强阿片受体结合的组合物及方法与流程
通过阿片类己二烯酸酯和任选取代的己二烯酸酯增强阿片受体结合的组合物及方法
1.本技术为2019年8月14日提交的第16/540,058号非临时美国专利申请的pct申请,并要求于2019年8月11日提交的名为“通过阿片类己二烯酸酯和任选取代的己二烯酸酯增强阿片受体结合的组合物及方法”的第62/885,311号先前美国临时专利的《美国专利法》第119条优先权。
技术领域
2.本发明涉及用于阿片受体调节相关治疗领域的阿片衍生组合物。
背景技术:
3.纳布啡(nubain)作为一种中度至重度疼痛镇痛药,于1979年推出,至今已有效应用于临床。它主要与麻醉药联合用于术前和术后镇痛,并在分娩时用于急性和慢性疼痛管理。最近,其用途已扩展到治疗运动障碍、皮肤病(瘙痒)和成瘾管理。
4.最近还表明,纳布啡可在慢性疼痛管理中预防阿片类药物的耐受性和依赖性。纳布啡是唯一一种不受《管制物质法》约束的麻醉性镇痛药,这证明其具有使用安全性。纳布啡的口服生物利用度较低。
5.已知的纳布啡前药旨在改善其药代动力学和药效动力学特性。韦氏词典将前药定义为一种药理非活性物质,即,在体内(通过酶促作用)转化成的药理活性药物的修饰形式。因此,franklin(wo 2010-gb52211)指出,可在酚羟基残基处对纳布啡进行修饰。
6.此外,纳布啡可偶联到氨基酸或短肽(wo 2011007247,a1)。也可使用二羧酸连接氨基酸和肽对纳布啡进行修饰(wo 2010112942,a1)。另外,也可使用氨基甲酸铵基团连接氨基酸和肽对纳布啡进行修饰(wo 2009092071,a2)。此外,jenkins(wo 2007022535,a2)指明,可在其酚或氮基团对纳布啡进行进一步的修饰。
7.wang指出,纳布啡可转化为酯前药(《控释杂志》,卷号:115,期号:2,页码:140-149,杂志,2006年)。flu(2005年1月11日,tw 226239,b)指出递送系统和纳布啡前药可提高其生物利用度。更具体地说,可提高纳布啡生物利用度的制剂包括植物油、共溶剂和有效量的纳布啡酯前药或其药学上可接受的盐。前药的一个目的是提高纳布啡的口服生物利用度,并延长纳布啡在体内的保留时间,从而保持较长的镇痛期,并降低镇痛成本。
8.hilfinger(us 20050137141,a1)指出纳布啡包含一种药物以及一种氨基酸(包含一个与所述药物连接的共价键)。huang(《国际制药学杂志》,卷号:297,期号:1-2,页码:162-171,杂志,2005年)指出了离子透入和电穿孔对经皮递送纳布啡(na)及其两种新型前药:纳布啡苯甲酸酯(nab)和癸二酰双那布扶林酯(sdn)(来自溶液以及水凝胶)的影响。
9.crooks(wo 2005009377,a2)指出,形成包含纳布啡的双前药可显著增加人体皮肤的药物经皮通量。uhrich(wo 2002009768,a2)指出了纳布啡的治疗性聚酯和聚酰胺。flu(ep 1149836,a1)指出了聚纳布啡衍生物的制备情况。pao(《色谱法杂志b辑:生物医学科学与应用》,卷号:746,期号:2,页码:241-247,杂志,2000年)指出了癸二酰双那布扶林酯的生
物利用度。
10.han(《国际制药学杂志》,卷号:177,期号:2,页码:201-209,杂志,1999年)指出了新型纳布啡前药控释用粘膜粘着颊片:制剂变量对药物释放和粘膜粘着性能的影响。sung(《国际制药学杂志》,卷号:172,期号:1-2,页码:17-25,杂志,1998年)指出了可生物降解聚合物基质中纳布啡前药的控释情况:前药亲水性和聚合物组合物的影响。yoa-pu(us 5750534,a)指出纳布啡酯具有长效镇痛作用。
11.shami(ep 85108258.6)指出,可将纳布啡进一步修饰为3-乙酰水杨酸。以下参考文献公开了其他纳布啡前药:us 6569449,b1;cn 1107333,a;ep 615756,a1;以及《国际制药学杂志》,卷号:38,期号:1-3,页码:199-209,杂志,1987年。
12.可通过各种递送系统来调节纳布啡、其药学上可接受的盐、酯或其前药的药代动力学和药效动力学特性。thus liu(《国际制药学杂志》,卷号:257,期号:1-2,页码:23-31,杂志,2003年)指出了纳布啡前药控释用可生物降解聚合物微球。sung(《欧洲药物科学杂志,卷号:18,期号:1,页码:63-70,杂志,2003年)指出了通过电穿孔经皮递送纳布啡及其前药的情况。fang(《药物研究》,卷号:51,期号:5,页码:408-413,杂志,2001年)指出了通过被动扩散和离子透入经皮递送纳布啡和纳布啡三甲基乙酸盐的情况。
13.必须区分提高口服生物利用度以及增加这些阿片类衍生物的阿片受体结合这两种情况。例如,此前据称,纳布啡苯氧基基团(例如,纳布啡的3-山嵛酸酯衍生物)(nb-39)的酯化作用已提高口服利用度。但是,当口服给药时,nb-39在大鼠和人类中所产生的累计镇痛作用弱于当量剂量的纳布啡。此外,在口服给药后,nb-39并未显著影响人体瞳孔放大(缩小),这表明阿片受体结合不良。
14.纳洛酮品牌名为“narcan”(及其他品牌名),是一种用于阻断阿片类药物作用(尤其是在过量情况下)的药物。为了减少阿片类药物滥用风险,也可联合施用纳洛酮与阿片类药物(在相同药片或化合物中)。例如,可向持续释放阿片类化合物的包衣中加入纳洛酮,防止持续释放化合物破碎,从而导致药物过量。
15.纳洛酮静脉内给药时,通常在两分钟内见效,注射到肌肉中时,在五分钟内见效。也可将其用作鼻腔喷雾剂。纳洛酮的作用通常持续约半小时至一小时。因此,可能需要多次施用纳洛酮,因为大多数阿片类药物作用的持续时间均大于纳洛酮。
16.对阿片类药物依赖个体施用纳洛酮可能引起阿片类药物戒断症状,例如,烦躁不安、躁动、恶心、呕吐、心率加快和出汗。为了防止这种情况,每隔几分钟施用小剂量的纳洛酮,直至达到预期效果。
17.在有既往心脏病史的个体或服用对心脏有负面影响的药物的个人中,还出现了进一步的心脏问题。已对有限数量的受试者施用纳洛酮,并进行了检测,结果表明,纳洛酮似乎在妊娠期很安全。
18.纳洛酮是一种非选择性、竞争性阿片受体拮抗剂。其通过逆转阿片类药物对中枢神经系统和呼吸系统的抑制作用来发挥作用。纳洛酮最初于1961年获得专利,并于1971年在美国批准用于阿片类药物过量治疗。
19.纳洛酮又名n-烯丙去甲羟吗啡酮或17-烯丙基-4,5a-环氧基-3,14-二羟基吗啡喃-6-酮,是一种合成吗啡喃衍生物,由羟吗啡酮(14-羟基二氢吗啡酮)(一种阿片类镇痛药羟吗啡酮)衍生而来,继而由吗啡(一种阿片类镇痛药,罂粟的天然成分)衍生而来。
20.纳洛酮是两种对映异构体(-)-纳洛酮(左旋纳洛酮)和(+)-纳洛酮(右旋纳洛酮)的外消旋混合物,仅前者在阿片受体处具有活性。所述药物高度亲脂,因此可快速穿透大脑,所达到的大脑-血清比远远大于吗啡。与纳洛酮相关的阿片类拮抗剂包括赛普罗定、纳美芬、nalodeine、纳洛醇和纳曲酮。
21.纳洛酮的化学半衰期如下:市售注射剂型和鼻用剂型的保质期分别为24个月和18个月。一项2018年的研究注意到,鼻用剂型和注射剂型的化学稳定期分别为36个月和28个月,这促使启动了一项尚未完成的五年稳定性研究。这表明,社区和医疗保健环境中贮藏的过期材料可能仍有效,远远超过其标签上的有效期。
22.一些关于阿片类拮抗剂的文章强调了目前已知制剂的缺点和问题以及对更稳定的修饰化合物(可安全用于阿片类药物成瘾患者)的需求。
23.adam bisaga的一篇名为“当芬太尼取代海洛因时,临床医生应该做什么?”的文章(发表于《成瘾》,第114卷,第781-86页,https://onlinelibrary.wiley.eom/doi/epdf/10.1111/add.14522)描述了高亲和性拮抗剂可能不足以阻断芬太尼的作用,可能需要更高的剂量,但这会带来全身安全问题。此外,芬太尼过量预防需要更高剂量的纳洛酮和重复给药,而芬太尼的过量预防窗口比海洛因短得多。
24.roger chou等人在名为“紧急医疗服务人员使用纳洛酮对疑似阿片类药物过量进行管理”(发表于《相对有效性评述》第193期,https://effectivehealthcare.ahrq.gov/sites/default/files/pdf/cer-193-naloxone-final_1.pdf)的文章中描述,现有纳洛酮给药指南可能不足以预防芬太尼和芬太尼类似物过量使用。
25.rachael rzasa lynn等人在名为“实现阿片类药物逆转的纳洛酮剂量:现有证据和临床意义”(发表于《药物治疗进展社会评论》,第9(1)卷,第63-88页,2018年,https://www.ncbi.nlm.nih.gov/pmc/articles/pmc5753997/pdf/10.1177_2042098617744161.pdf)的文章中描述,向芬太尼麻醉患者施用双倍剂量的纳洛酮时,摄氧量没有改善,而施用四倍剂量的纳洛酮时,有明显改善。此外,他还指出,阿片类激动剂与mu阿片受体之间的相互作用可能是从许多阿片类药物的呼吸作用中恢复速度的最大决定因素,可能不会随着纳洛酮剂量的增加而显著加速,而是对最小有效剂量有反应,而对于丁丙诺啡等化合物来说,更高剂量的纳洛酮甚至可能失效。然后,他引用了大量报告,其中描述了芬太尼过量最初对鼻内施用纳洛酮无反应,静脉注射纳洛酮(如有)仅实现了短暂逆转,需要另外进行静脉注射或连续输注,以防毒性和呼吸抑制复发。
26.ia.elkiweri等人在名为“p-糖蛋白和有机负离子转运蛋白的竞争性底物不同程度地减少了芬太尼和洛派丁胺的血液器官运输:在sprague-dawley大鼠中的药代动力学和药效动力学”(2009年在线发表于https://www.ncbi.nlm.nih.gov/pubmed/19095843)的文章中描述,纳洛酮和芬太尼共享一个细胞流入转运蛋白,由于血浆中芬太尼的浓度较高,此转运蛋白饱和,因此,无论剂量如何,纳洛酮均无法快速流过bbb。
27.rebecca mcdonald等人在名为“浓缩纳洛酮鼻腔喷雾剂在阿片类药物过量逆转方面的药代动力学:第i阶段健康志愿者研究”(发表于《成瘾》,第113卷,第484-93页)的文章中描述,高浓度2mg纳洛酮鼻内(i.n.)喷雾剂的早期吸收率与肌肉内(i.m.)0.4mg注射类似,可用作家用解毒剂。他表明,在不存在过度拮抗作用风险的情况下,可鼻内施用高剂量的纳洛酮。
28.jiten ranchhodbhai patel等人公开了(第wo 2013093931号出版物-申请pct/in20 12/00590,于2012年9月6日提交)一种包含纳洛酮氨基甲酸酯的新酰肼基。
29.baohua huang等人在名为“基于吲哚醌的纳洛酮前药的人血浆介导缺氧活化”(2009年发表于《生物有机化学与医药化学通讯》,19(17),5016-5020)的文章中描述,基于吲哚醌的纳洛酮前药可逆转阿片类药物诱导缺氧。
30.i.ukrainets等人在出版物《杂环化合物化学》(2009年,45(4),第405-416页)中公开了将纳洛酮3-o-酰基衍生物作为其潜在前药的研究。
31.xuemei peng等人在名为“包含在μ、δ和k阿片受体处与纳布啡和纳洛酮相连的布托啡诺的二价配体的药理学特性”(发表于《药物化学杂志》(2007年5月),50(9),2254-2258)的文章中公开了包含与纳洛酮相连的布托啡诺的二价配体。
32.i.romanov等人在俄罗斯专利出版物(ru 2221566-于2004年1月20日发表)中描述,可将n-取代14-羟基吗啡喃酯用作高效低毒抗复发剂,在单次皮下或肌肉内注射后,可产生长期阿片类药物保护作用。
33.i.romanov等人在俄罗斯专利出版物(ru 2215741-于2003年11月10日发表)中描述了n-取代14-羟基吗啡喃酯的制备方法。
34.euro-celtique,s.a.,chevchuk等人在第wo 2003070191号专利出版物(pct/us/2003/004999-于2003年8月28日发表)中描述了使用装有3-酰基-取代拮抗剂的防破坏皮肤器械预防疼痛的方法。
35.lu zhengtang在第cn 1204649号中国专利(于1999年1月13日发表)中公开了纳洛酮酯的制备方法。
36.s.lazar等人在名为“纳洛醇和纳曲酮的磷酸酯和硫酸酯的合成和生物活性”(发表于《欧洲药物化学杂志》(1994年),第29(1)卷,第45-53页)的文章中描述了纳洛酮的磷酸酯和硫酸酯的合成和生物活性。
37.hussein等人描述(《药物研究》(1988年),第5(9)卷,第615-18页),各种纳洛酮前药(3-苯氧基已酯化)均无苦味,在狗中的颊生物利用度更高。
38.elie gabriel shami在第ep 170090号欧洲专利出版物中描述了3-羟基吗啡喃的苯甲酸酯前药衍生物。上述出版物均通过本说明书的引用,成为本说明书的一部分。
39.所引用的这些出版物均未描述纳洛酮与分子内所含己二烯酸酯联合使用的情况,也未表明在向个人施用时,此类分子将导致并提供实质上更有效、更持久的中和/清醒作用。
技术实现要素:
40.本发明涉及一种阿片类药物及其拮抗剂的新型修饰物,其可增加口服时的阿片受体结合。更具体地说,本发明涉及合适的阿片受体调节剂(例如,纳布啡、丁丙诺啡、氢吗啡、吗啡、喷他佐辛、布托啡诺、纳洛酮等)或相关化合物的修饰物,其可改善口服给药时阿片类药物与阿片受体的结合。
41.本发明进一步涉及将阿片类药物用于(但不限于)以下病症时缓解阿片类药物低口服生物利用度问题的方法:疼痛管理、姑息护理、麻醉(例如,术后)、皮肤病(例如,瘙痒)、成瘾(脱瘾或管理)、某些运动障碍(例如,帕金森氏病左旋多巴诱发异动症(lid)、与图雷特
综合征相关的运动障碍、迟发性运动障碍和亨廷顿病)等。
42.本发明涉及一种阿片类药物(例如,纳布啡)的新型修饰物,其可提供意想不到的结果,即,增加口服给药时阿片受体的结合。因此,这种新型修饰物提供优质护理,适用于各种治疗适应症,包括需要口服阿片类药物的慢性病。
43.本发明涉及一种阿片类拮抗剂的新型修饰物,如与分子内所含己二烯酸酯联合使用的纳洛酮,在向个人或患者给药时,将提供实质上更有效、更持久的中和/清醒作用。
44.将参照以下附图,对本发明的新特征进行进一步说明。
附图说明
45.本专利或申请文件包含至少一个彩色附图。根据请求,在支付必要费用后,专利局将提供本专利或专利申请出版物的副本(包含彩色附图)。
46.图1显示了根据本发明至少一个实施例配制的nb-20化合物的nmr1h谱。
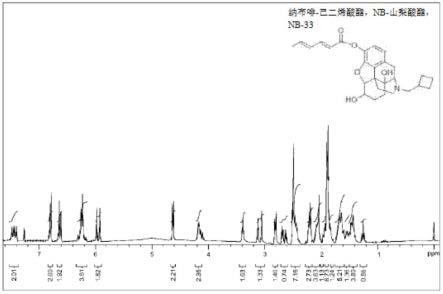
47.图2显示了根据本发明至少一个实施例配制的nb-33化合物的nmr1h谱。
48.图3显示了根据本发明至少一个实施例配制的nb-39化合物的nmr1h谱。
49.图4显示了根据本发明至少一个实施例配制的nb-51化合物的nmr1h谱。
50.图5显示了根据本发明至少一个实施例配制的nb-52化合物的nmr1h谱。
51.图6显示了根据本发明至少一个实施例配制的nb-56化合物的nmr1h谱。
52.图7显示了根据本发明至少一个实施例配制的nb-58化合物的nmr1h谱。
53.图8显示了根据本发明至少一个实施例配制的nb-78化合物的nmr1h谱。
54.图9a显示了最受欢迎的纳布啡构象异构体(与共晶化配体β-fna叠加)的结合模式和分子间相互作用。
55.图9b显示了最受欢迎的纳洛酮构象异构体(与共晶化配体β-fna叠加)的结合模式和分子间相互作用。
56.图10a显示了4dkl结合位点中最受欢迎的nx-90构象异构体的结合模式和分子间相互作用。
57.图10b显示了4dkl结合位点中最受欢迎的nb-33构象异构体的结合模式和分子间相互作用。
58.图10c显示了与met 151的分子间相互作用(如nb-33构象异构体所示),其结合模式与最受欢迎的构象异构体相似。
59.图10d显示了4dkl结合位点中最受欢迎的nb-39构象异构体的结合模式和分子间相互作用。
60.图11a显示了最受欢迎的纳布啡(黄色)、纳洛酮(粉色)构象异构体以及在4dkl阿片类结合位点中叠加的共晶化β-fna(白色)。
61.图11b显示了最受欢迎的nx-90(蓝色)、nb-33(红色)、nb-39(青色)构象异构体以及在4dkl阿片类结合位点中叠加的共晶化β-fna(白色)。
62.图12a-图12c显示了在4dkl结合位点分子表面上与至少一个实施例中nx-90、nb-33和nb-39对接构象异构体分别接触的疏水性(红色)和亲水性(黄色)接触偏好区。
63.图13包含图表1,其中显示,与母体阿片类药物nb的等摩尔剂量相比,至少一个实施例中nb-33具有优越的镇痛特性。
具体实施方式
64.本发明包括形成用于阿片受体调节相关治疗领域的阿片衍生组合物,其包含单分子中的己二烯酸酯和阿片残基。
65.参照表1对本发明及组合物的各个方面和特征进行了说明,表1显示了化合物nb、nb-20、nb-28、nb-31、nb-32、nb-33、nb-39、nb-46、nb-51、nb-52、nb-56、nb-58、nb-76、nb-78的所选特性。
66.根据本发明至少一个实施例配制的所选化合物(示例包括nb-20、nb-33、nb-39、nb-51、nb-52、nb-56、nb-58、nb-78)的nmr 1h谱示例分别如图1-8所示。
67.令人惊奇的是,与母体阿片类药物化合物相比,根据本发明至少一个实施例制备的阿片类药物的3-己二烯酸酯衍生物可增加阿片受体结合。因此,当口服给药时,纳布啡3-己二烯酸酯(nb-33)在大鼠和人类中所产生的镇痛作用优于当量剂量的纳布啡3-山嵛酸酯(nb-39)和纳布啡(nb)。此外,在人类中观察到nb-33对瞳孔放大(缩小)产生显著影响,这表明阿片受体结合极佳。
68.出乎意料的是,当研究本发明至少一个实施例的作用时,发现氧基基团酯不饱和位点的位置和数量是己二烯骨架独有的,对于形成阿片受体的最佳结合是所需条件。因此,纳布啡3-烷烃酸酯(例如,nb-33)的镇痛作用优于母体阿片类药物,而纳布啡的其他不饱和酸衍生物(例如,nb-31、nb-32、nb-52或nb-78)在大鼠中则不会产生镇痛作用。
69.此外,对本发明的至少一个实施例进行评价,结果发现,纳布啡3-己二烯酸酯具有独特而鲜明的阿片受体特征,并在细胞中表达人重组阿片受体。
70.根据至少一个实施例,本发明所述的化合物包含通式i或其药学上可接受的盐
[0071][0072]
其中,r1、r2、r3、r4或r5选自h、任选取代的c1-3和oalk,双键具有e或z几何形状,y为阿片残基。
[0073]
在至少一个实施例中,本发明进一步涉及将阿片类药物用于(但不限于)以下病症时缓解阿片类药物低口服生物利用度问题的方法:疼痛管理、姑息护理、麻醉(例如,术后)、皮肤病(例如,瘙痒)、成瘾(脱瘾或管理)、某些运动障碍(例如,帕金森氏病左旋多巴诱发异动症(lid)、与图雷特综合征相关的运动障碍、迟发性运动障碍和亨廷顿病)等。
[0074]
在至少一个实施例中,本发明涉及一种合适的阿片受体调节剂或相关化合物苯氧基基团修饰物的任选取代的己二烯酸酯,用于改善口服给药时阿片类药物与阿片受体的结合。
[0075]
在另一个实施例中,本发明涉及一种合适的阿片受体调节剂(包括但不限于氢吗啡、吗啡、纳布啡、喷他佐辛、布托啡诺、丁丙诺啡、纳洛酮)或相关化合物3-苯氧基基团修饰物的任选取代的己二烯酸酯,用于改善口服给药时阿片类药物与阿片受体的结合。
[0076]
在至少另一个实施例中,本发明涉及一种合适的阿片受体调节剂或相关化合物的3-己二烯酸酯修饰物,用于改善口服给药时阿片类药物与阿片受体的结合。
[0077]
在至少一个实施例中,本发明涉及一种纳布啡或其药学上可接受的盐的3-己二烯
酸酯修饰物,用于改善口服给药时的阿片受体结合。
[0078]
在另一个实施例中,本发明涉及一种纳布啡或其药学上可接受的盐的3-己二烯酸酯修饰物,用于改善口服给药时的疼痛管理质量。
[0079]
在另一个或多个实施例中,本发明涉及一种纳布啡或其药学上可接受的盐的3-己二烯酸酯修饰物,用于改善静脉内、鼻内、经皮、舌下、直肠、局部、肌肉内、皮下或吸入给药时的疼痛管理质量。
[0080]
下面提供了根据本发明至少一个实施例制备的化合物的进一步示例。示例1中每个化合物的化学名称、组成和代号如下表1所示。
[0081]
示例1
[0082]
(e)-3-(环丁烷甲基)-9-((3,7-二甲基辛-2,6-二亚乙基三胺-1-基)氧基)-1,2,3,4,5,6,7,7a-八氢-4ah-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-4a,7-二醇,纳布啡-香叶基,(nb-20)。在室温下,在盐酸纳布啡(400mg,1.0mmol)的丙酮(20ml)和甲苯(20ml)悬浮液中加入碳酸氢钾(280mg,2.0mmol)。加入香叶基溴(320mg,1.5mmol)。在反流条件下将反应混合物搅拌4小时,并在室温下过夜搅拌。蒸发反应混合物,并通过柱色谱法(硅胶,etoac/庚烷/甲醇,1:1:0.10)纯化残留物。蒸发所选部分后,形成无色油,通过hplc确定,收率为45%,纯度为91%。通过nmr 1
h确认结构。
[0083]
3-(环丁烷甲基)-9-(((2e,6e)-3,7,11-三甲基十二烷基-2,6,10-三亚乙基四胺-1-基)氧基)-1,2,3,4,5,6,7,7a-八氢-4ah-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-4a,7-二醇,纳布啡-法尼基,(nb-28)。根据nb-20的程序制备此化合物,用法尼基溴代替香叶基溴。通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化粗料。蒸发所选部分后,获得无色油,通过hplc确定,收率为53%,纯度为93%。通过nmr 1
h确认结构。
[0084]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基十一-10-烯酸酯,纳布啡-十一烯酸酯,(nb-31)。在0℃温度下,在十一碳烯酸(1.0g,5.4mmol)的thf(30ml)溶液中加入edci(1.04g,5.4mmol),同时不断搅拌。将反应混合物搅拌10分钟,在0℃温度下,加入盐酸纳布啡(2.13g,5.4mmol)、三甲胺(1.1g,10.9mmol)和4-二甲氨基吡啶(0.22g,1.8mmol)。在0℃温度下继续搅拌1小时,并在室温下过夜搅拌。过滤反应混合物,蒸发滤液,并通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化残留物。蒸发所选部分后,形成白色固体,通过hplc确定,收率为78%(2.2g),纯度为95%。通过nmr 1
h确认结构。
[0085]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基(e)-3,7-二甲基辛-2,6-二烯酸酯,纳布啡-香叶酸酯,(nb-32)。根据nb-31的程序制备此化合物,用香叶酸代替十一碳烯酸。通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化粗料。蒸发所选部分后,形成白色固体,通过hplc确定,收率为67%,纯度为96%。通过nmr 1
h确认结构。
[0086]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基(2e,4e)-2,4-己二烯酸酯,纳布啡-山梨酸酯,(nb-33)。在0℃温度下,在己二烯酸(0.68g,6.1mmol)的thf(30ml)溶液中加入edci(1.16g,6.1mmol),同时不断搅拌。将反应混合物搅拌10分钟,在0℃温度下,加入盐酸纳布啡(2.39g,6.1mmol)、三甲胺(1.2g,12mmol)和4-二甲氨基吡啶(0.25g,2mmol)。在0℃温度下继续搅拌1小时,并在室
温下过夜搅拌。过滤反应混合物,蒸发滤液,并通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化残留物。蒸发所选部分后,形成白色晶体,通过hplc确定,收率为75%(2.05g),纯度为98%。通过nmr 1
h确认结构。
[0087]
3-(环丁烷甲基)-9-(((2e,4e)-己-2,4-二烯酰)氧基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-3-鎓氯化物,纳布啡-山梨酸酯,盐酸,(nb-56)。在0℃温度下,将hcl(气体)吹入纳布啡-山梨酸酯(nb-33)(0.4g,0.89mmol)的mtbe(15ml)溶液中。将反应混合物搅拌1小时,过滤固体,用mtbe进行洗涤,并真空干燥。通过hplc确定,收率为81%(0.35g),纯度为98%。通过nmr1h确认结构。
[0088]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基山嵛酸酯,纳布啡-山嵛酸酯,(nb-39)。在0℃温度下,在山萮酸(1.0g,2.9mmol)的thf(50ml)溶液中加入edci(0.56g,2.9mmol),同时不断搅拌。将反应混合物搅拌30分钟,在0℃温度下,加入盐酸纳布啡(1.16g,2.9mmol)、三甲胺(0.29g,2.9mmol)和4-二甲氨基吡啶(0.12g,1.0mmol)。在0℃温度下继续搅拌1小时,并在室温下过夜搅拌。过滤反应混合物,蒸发滤液,并通过柱色谱法(硅胶,etoac/庚烷,1:2)纯化残留物。蒸发所选部分后,形成白色固体,通过hplc确定,收率为73%(1.45g),纯度为97%。通过nmr 1
h确认结构。美国专利5750534也对nb-39的合成和特性进行了说明。
[0089]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基异丁酸酯,纳布啡-异丁酸酯,(nb-46)。根据nb-31的程序制备此化合物,用异丁酸代替十一碳烯酸。通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化粗料。蒸发所选部分后,形成白色晶体,通过hplc确定,收率为54%,纯度为95%。通过nmr 1
h确认结构。
[0090]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基3-甲基丁-2-烯酸酯,纳布啡-3,3-二甲基丙烯酸酯,(nb-51)。根据nb-31的程序制备此化合物,用3.3-二甲基丙烯酸代替十一碳烯酸。通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化粗料。蒸发所选部分后,形成白色晶体,通过hplc确定,收率为77%,纯度为95%。通过nmr 1
h确认结构。
[0091]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基(e)-2-甲基丁-2-烯酸酯,纳布啡-2,3-二甲基丙烯酸酯,(52)。根据nb-31的程序制备此化合物,用2.3-二甲基丙烯酸代替十一碳烯酸。通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化粗料。蒸发所选部分后,形成白色晶体,通过hplc确定,收率为75%,纯度为96%。通过nmr 1
h确认结构。
[0092]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基2-甲基丁-2-烯酸酯,纳布啡-2-甲氧基巴豆酸酯,(nb-58)。根据nb-31的程序制备此化合物,用2-甲氧基-巴豆酸代替十一碳烯酸。通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化粗料两次。蒸发所选部分后,形成白色油,通过hplc确定,收率为27%,纯度为94%。通过nmr 1
h确认结构。
[0093]
7-乙酰氧基-3-(环丁烷甲基)-4a-羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基(2e,4e)-2,4-己二烯酸酯,纳布啡-己二烯酸酯-乙酸酯,(nb-76)。在40-50℃温度下,在乙酸酐(7.0ml)中过夜搅拌nb-33(0.5g,1.1mmol)。加入etoh
(20ml),蒸发反应混合物。通过柱色谱法(硅胶,etoac/庚烷,1:2)纯化残留物两次。蒸发所选部分后,形成白色晶体,通过hplc确定,收率为50%(1.45g),纯度为97%。通过nmr 1
h确认结构。
[0094]
3-(环丁烷甲基)-4a,7-二羟基-2,3,4,4a,5,6,7,7a-八氢-1h-4,12-甲桥苯并呋喃并[3,2-e]异喹啉-9-基肉桂酸酯,纳布啡-肉桂酸酯,(nb-78)。根据nb-31的程序制备此化合物,用2-反式-肉桂酸代替十一碳烯酸。通过柱色谱法(硅胶,etoac/庚烷,1:1)纯化粗料。蒸发所选部分后,形成白色晶体,通过hplc确定,收率为67%,纯度为94%。通过nmr 1
h确认结构。
[0095]
表1
[0096]
[0097]
[0098]
[0099]
[0100]
[0101]
[0102][0103]
示例2
‑‑
在模拟胃肠液(sgif)中的稳定性。
[0104]
nb-33在模拟胃肠液(sgif)中的稳定性评估如下,表1汇总了各项化合物数据。
[0105]
sgif为0.5%胃蛋白酶(阿法埃莎公司,胃蛋白酶,猪胃)的0.1n hcl水溶液。将每种衍生物(50mg)与sgif(50ml)混合,并在37℃温度下,在摇床上进行培养。在t=0小时、0.5小时、1小时、2小时和4小时时,通过hplc监测纳布啡的水解和释放情况。可接受标准为在4小时后不少于80%的衍生物仍完好无损。
[0106]
示例3-在人血浆中的稳定性。
[0107]
nb-56在人血浆中的稳定性评估如下,表1汇总了各项化合物数据。
[0108]
在10ml血浆(混合正常人血浆,柠檬酸钠,创新研究公司)中溶解nb-56(1.0mg),同时在20℃温度下搅拌10分钟。在37℃温度下对溶液进行培养。每份试样取1ml溶液。在样本溶液中加入mecn(0.05ml)。摇晃1分钟,然后进行离心(15分钟,14.000r/m)。滤出并使用etoac(2
×
20ml)提取上清液。使用mgso4对混合提取物进行干燥,并进行真空浓缩。在甲醇(20μl)中溶解残留物。将溶液用于hplc进样。
[0109]
在t=0小时、0.5小时、1小时、2小时和4小时时,通过hplc监测纳布啡的水解和释放情况。可接受标准为在4小时后不少于20%的衍生物发生水解。
[0110]
示例4
[0111]
表2-nb-33的人重组阿片受体数据
[0112][0113]
使用在cho-k1细胞中表达的人重组阿片受体(μ、κ或δ)。在37℃温度下,在修饰hbss ph 7.4缓冲液中将受试化合物(nb-33)/或运载体与细胞(4
×
10e5/ml)培养30分钟。通过tr-fret对反应的camp水平进行评估。由欧陆药物发现服务公司筛选0.3um、1um和3um的化合物。
[0114]
表2汇总了化合物nb-33的数据。
[0115]
示例5
[0116]
使用纳布啡、nb-31、nb-32、nb-33、nb39、nb-51、nb-52、nb-76和nb-78对sprague-dawley大鼠进行试验。
[0117]
将三十只sprague-dawley大鼠(12周龄;雄性)随机分配为10组,通过管饲法对每组进行以下一种治疗:1.芝麻油;2.纳布啡(在芝麻油中;60um/kg);3.nb-31(在芝麻油中;60um/kg);4.nb-32(在芝麻油中;60um/kg);5.nb-33(在芝麻油中;60um/kg);6.nb-39(在芝麻油中;60um/kg);7.nb-51(在芝麻油中;60um/kg);8.nb-52(在芝麻油中;60um/kg);9.nb-76(在芝麻油中;60um/kg);10.nb-78(在芝麻油中;60um/kg)。每只大鼠仅口服一次药物。
[0118]
通过冷乙醇甩尾试验评估抗伤害性感受活性(如《麻醉与镇痛》;2003年;97;806-9所述)。将试验温度设置为-20℃,截止时间为40秒。在给药前t=0时,立即对所有大鼠进行试验。在口服后t=0小时、0.25小时、0.5小时、1小时、1.5小时、2小时、3小时和5小时,测量盐水、纳布啡及纳布啡衍生物的抗伤害性感受阈值。
[0119]
表1最后一列所示的数据表明nb-33的结果良好。
[0120]
示例6
[0121]
健康志愿者口服nb-33抗伤害性感受作用的双盲nb盐酸和nb-39对照试验。向三名健康志愿者各分配包括如下6粒不透明明胶胶囊的一组药物:2
×
nb盐酸(mw=393.4;39mg)、2
×
nb-33(mw=451.6;45mg)和2
×
nb-39(mw=680.0;68mg)。每名健康志愿者每周从所述分配组中随机获得一粒药,并口服。在口服后t=0小时、0.25小时、0.5小时、1小时、1.5小时、2小时、3小时和5小时,测量热痛阈值(50℃热水)以及瞳孔缩小情况。
[0122]
在一周的洗脱期后,每名志愿者重复执行所述方案,直至口服所述分配组中的所有药物。表2和表3分别显示了热痛阈值%mpe=[(试验潜伏期-基线潜伏期/(基线潜伏期)]
×
100以及瞳孔缩小%mpe=[(试验直径)-基线直径/(基线直径)]
×
100的各项数据。
[0123]
表3、表4和表5a-d显示,口服时,nb-33的镇痛和瞳孔缩小作用优于母体阿片类药
物nb和母体阿片类前药nb-39。如表5a-d所示,镇痛和瞳孔缩小作用差异具有统计学意义。
[0124]
表3
[0125][0126][0127]
表4
[0128][0129]
通过独立样本t检验来比较两对样本的%mpe镇痛和瞳孔缩小平均值:
[0130]
nb-33和nb以及nb-33和nb-39。使用spss(v.25)进行所有分析。
[0131]
*粗体表示在a=0.05时具有统计学意义。
[0132]
表5a-d显示了nb-33与nb和nb-39之间的镇痛和瞳孔缩小作用比较情况。
[0133][0134][0135]
以下表6a和图13中的图表1显示了nb-33在randall-selitto大鼠上的其他试验结果,证明了其相对于基础化合物的疗效和益处(包括稳定性更高)。
[0136]
以下表6a和图13中的图表1显示了nb-33在randall-selitto大鼠上的其他试验结果,证明了其相对于基础化合物的疗效和益处(包括稳定性更高)。
[0137]
表6a
[0138]
wo#10656913 ab137003
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
物种/品系/性别:比率
[0139][0140][0141]
图13中图表1上的数据显示,nb-33的镇痛特性优于等摩尔剂量的母体阿片类药物nb。图13以图表形式显示了1310nb-33(在图13中标记为1310)相对于基础nb化合物nb(标记为1320)的结果。
[0142]
示例7
[0143]
本发明以下列化合物(如下所示)为示例,但不限于下列化合物。下列化合物(如以
下表7所示)提供了由至少一个实施例所述己二烯酸酯进行修饰的各种阿片类药物的非限制性实施例。
[0144]
表7
[0145]
[0146]
[0147]
[0148]
[0149]
[0150]
[0151]
[0152][0153]
示例8-纳布啡/纳洛酮阿片类拮抗剂与μ阿片受体的分子对接。
[0154]
从rcsb蛋白数据库(pdb条目:4dkl,https://www.rcsb.org/structure/4dkl)中下载人m阿片受体晶体结构。使用moe对接程序(moe模拟模块2014.0901的一部分)进行硅片筛选。根据等式
△
g=rtin(ki)计算解离常数(ki),式中,
△
g表示结合自由能,等于gbvi/wsa dg评分函数,r为气体常数,t为温度。从固定温度(300k)下的结合自由能开始计算ki。
[0155]
拮抗剂纳布啡和纳洛酮显示了asp 147与其铵基之间的关键相互作用。众所周知,与asp 147的这种结合对于大多数已知阿片类激动剂/拮抗剂来说属于一种典型情况。纳布啡和纳洛酮的其他重复相互作用为附着于芳基环(3位)的羟基与水分子结合,有助于稳定阿片受体的非活性状态。与纳布啡不同,附着于叔碳原子(14位)的纳洛酮羟基参与其他asp 147氢键结合活动。
[0156]
图9a显示了最受欢迎的纳布啡构象异构体(与共晶化配体β-fna叠加)的结合模式和分子间相互作用。图9b显示了最受欢迎的纳洛酮构象异构体(与共晶化配体β-fna叠加)
的结合模式和分子间相互作用。
[0157]
在纳布啡和纳洛酮中,附着于芳基环(3位)的羟基与水分子结合,有助于稳定阿片受体的非活性状态。与纳布啡不同,附着于叔碳原子(14位)的纳洛酮羟基参与其他asp 147氢键结合活动。
[0158]
图10a显示了4dkl结合位点中最受欢迎的nx-90构象异构体的结合模式和分子间相互作用。
[0159]
图10b显示了4dkl结合位点中最受欢迎的nb-33构象异构体的结合模式和分子间相互作用。
[0160]
图10c显示了与met 151的分子间相互作用(如nb-33构象异构体所示),其结合模式与最受欢迎的构象异构体相似。
[0161]
图10d显示了4dkl结合位点中最受欢迎的nb-39构象异构体的结合模式和分子间相互作用。
[0162]
图11a显示了最受欢迎的纳布啡(黄色)、纳洛酮(粉色)构象异构体以及在4dkl阿片类结合位点中叠加的共晶化β-fna(白色)。图11b显示了最受欢迎的nx-90(蓝色)、nb-33(红色)、nb-39(青色)构象异构体以及在4dkl阿片类结合位点中叠加的共晶化β-fna(白色)。
[0163]
nx-90、nb-33、nb-39的计算解离常数(ki)表明,对m受体的亲和性高于(nb-33、nb-39)或低于(nx-90)纳布啡和纳洛酮的亲和性。与最受欢迎的纳洛酮和纳布啡构象异构体相似,nx-90、nb-33和nb-39保留了与asp147残基的关键氢键结合。在这种对接模式下,将附着于氮原子的“消息”递送给m受体结合袋精确区域上的正确“地址”。但是,与已知m拮抗剂(例如,纳布啡和纳洛酮;图11a)的结合模式不同,在结合位点将nx-90、nb-33和nb-39的刚性框架旋转180
°
(图11b)。相反,描述纳布啡和纳洛酮结合的结合模式不可用于nb-33、nb-39和nx-90的所有计算构象异构体。
[0164]
此外,nx-90和nb-33通过附着于叔碳原子(14位)的羟基与met 151发生独特氢键结合。这种相互作用使nx-90和nb-33不同于nb-39,即,环己烷片段处(6位)的羟基与lys a233形成氢键。nx-90和nb-33的第二个区别因素为己二烯酸残基的刚性偶联系统具有特别的疏水性柱状分子表面。同时,残基cys217、thr218、asn127、gln124、trp133、leu219在结合袋内的己二烯基“尾巴”周围建立额外的互补疏水性分子表面(图12a和图12b),而在高柔性且缺乏偶联的二十二酰基“尾巴”周围的结合位点区域则不存在可辨别的疏水性表面(图12c)。
[0165]
图12a-图12c显示了在4dkl结合位点分子表面上与nx-90(如图12a所示)、nb-33(如图12b所示)和nb-39(如图12c所示)对接构象异构体分别接触的疏水性(红色)和亲水性(黄色)接触偏好区。
[0166]
这些示例(尤其是图9-图12中的示例)证实了本发明的至少一个特征,即,使用包含至少两个偶联双键的亲脂性基团修饰阿片类药物可改善与阿片受体的相互作用。
[0167]
这些示例还证实了本发明的另一个特征,即,通过在活性位点将阿片类药物旋转180
°
并创建与受体的其他相互作用模式(包括独特的疏水袋),使用包含至少两个偶联双键的亲脂性基团修饰阿片类药物可改善与阿片受体的相互作用。
[0168]
这些示例进一步证实了本发明的另一个特征,即,使用包含至少两个偶联双键的
亲脂性基团修饰阿片类药物可至少改变本发明某些实施例中的阿片类药物特性。
[0169]
这些示例还证实了本发明的另一个特征,即,使用包含至少两个偶联双键的亲脂性基团修饰阿片类药物可至少改善本发明某些实施例中的阿片类药物拮抗特性。
[0170]
阿片受体拮抗剂的性能得到改善
[0171]
如上所述,除阿片类药物的性能和镇痛质量得到改善之外,本发明还包括至少一个实施例,其中表明,己二烯酸酯还改善了阿片受体拮抗剂(如纳洛酮)的性能。
[0172]
在至少一个实施例中,本发明(尤其是己二烯酸酯)与纳洛酮组或化合物中的至少一种(或多种)结合并进行试验。
[0173]
在本发明的至少一个实施例中,纳洛酮(包含己二烯酸酯)包含在分子中,在向受试者施用时,将提供实质上更有效、更持久的中和/清醒作用。
[0174]
在本发明的至少一个实施例中,合成并分析采用以下化学式的化合物nx-90和nx-97(如以下表8所示)。
[0175]
表8
[0176][0177]
向受试者施用时,可能注意到清醒作用。可根据本发明的至少一个实施例使用并合成这种化合物,称为纳洛酮-山梨酸酯(nx-90)。根据至少一个实施例制备化合物的程序
如下。
[0178]
在0℃温度下,在己二烯酸(0.74g,6.61mmol)的thf(50ml)溶液中加入edci.hcl(1.36g,7.12mmol),同时不断搅拌。加入三乙胺(1.39g,13.8mmol)。在0℃温度下搅拌2小时。在0℃温度下,加入盐酸纳洛酮(2.00g,5.5mmol)和4-二甲氨基吡啶(0.10g,0.82mmol)。在0℃温度下继续搅拌1小时,并在室温下过夜搅拌。过滤反应混合物,蒸发滤液,并通过柱色谱法(硅胶,etoac/庚烷/三乙胺,2:1:0.5%)纯化残留物两次。蒸发所选部分后,形成白色晶体,通过hplc确定,收率为32%(0.75g),纯度为98%。通过nmr 1
h确认结构。
[0179]
对nx-90化合物的特性进行了研究,获得并证实了以下结果和具体益处(包括稳定性数据)。
[0180]
表9-nx-90(批号:α-1-91)稳定性(gif)(由alfacheminvent公司检测)。
[0181][0182][0183][0184]
根据结果和观察,如表9所示,与知名药物纳洛酮相比,nx-90改善明显。
[0185]
下表11进一步显示了nb-33或相似化合物与不同阿片类药物以及nx-90与本发明至少一个实施例所述阿片类拮抗剂的组合示例。
[0186]
有充分文献证实且众所周知,阿片类药物可用于治疗以下病症:疼痛管理、姑息护理、术后麻醉、皮肤病、成瘾、运动障碍、帕金森氏病左旋多巴诱发异动症(lid)、与图雷特综合征相关的运动障碍、迟发性运动障碍和亨廷顿病等。用于治疗这些病症的阿片类药物的效力和有效性会影响治疗的成功程度。
[0187]
相应地,阿片受体结合越好,根据本发明至少一个实施例治疗所述病症的结果越有效。例如,因为其阿片受体结合更好,使用己二烯酸酯进行修饰的阿片类药物在治疗上述病症时更有效。
[0188]
因此,在本发明的至少一个实施例中,根据本发明配制的一种合成化合物(如nb-33或nx-90等)可用于治疗以下一种病症:疼痛管理、姑息护理、麻醉(例如,术后)、皮肤病(例如,瘙痒)、成瘾(脱瘾或管理)和/或运动障碍(例如,帕金森氏病左旋多巴诱发异动症(lid)、与图雷特综合征相关的运动障碍、迟发性运动障碍和亨廷顿病)。
[0189]
示例9
[0190]
表10显示了nx 90的人重组阿片受体数据。
[0191][0192][0193]
使用在cho-k1细胞中表达的人重组阿片受体(μ、κ或δ)。在37℃温度下,在修饰hbss ph 7.4缓冲液中将受试化合物((nx-90)/或运载体与细胞(4
×
10e5/ml)培养30分钟。通过tr-fret对反应的camp水平(camp和/或钙通量)进行评估。由欧陆药物发现服务公司筛选0.1um、0.3um和1um的化合物。
[0194]
表10汇总了化合物nb-90的数据。表中显示,nx-90不属于药理学惰性化合物,并且具有其独特的阿片类特征,与纳洛酮的药理学特征类似。另外,表中显示,nb-33不属于药理学惰性化合物,并且具有独特的阿片类特征,与nb的药理学特征类似。
[0195]
这些结果非常令人吃惊,因为本领域认为,3-苯氧基位置(包含脂肪酸)的此类修饰物(例如,nb-39)属于前药,并且根据定义,属于药理学惰性化合物。根据本发明的至少一个实施例,nx-90和nb-33不具有药理学惰性,不属于前药。
[0196]
在所有情况下,应理解,上述示例和化合物仅用于说明代表本发明应用的许多可能特定实施例。在不脱离本发明的精神和范围的情况下,可根据本发明的原则轻松设计各种其他布置。
[0197]
表11
[0198][0199]
[0200]
[0201]
[0202]
[0203]
[0204]
[0205]
[0206]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1