包含西苯唑啉或其盐的药物制剂的制作方法
1.本发明涉及包含西苯唑啉或其盐的药物组合物,并涉及包含西苯唑啉或其盐的药物制剂,其中药物制剂包括速释层和缓释层。
背景技术:
2.西苯唑啉(cifenline,2-(2,2-二苯基环丙基)-4,5-二氢-1h-咪唑,53267-01-9(cas号))是一种具有以下化学式1结构的化合物,并且是一种用作抗心律失常剂的药物(1族钠通道阻断剂)。此外,西苯唑啉作为钙通道阻滞剂和钾通道阻滞剂的作用也是已知的。
[0003][0004]
由于西苯唑啉半衰期短,在药物被人体吸收后,西苯唑啉会迅速消失,因此,由于其药效持续时间短,应每天给药几次。西苯唑啉目前已以治疗心律失常的片剂形式开发并上市,每天给药三次。
[0005]
因此,需要开发一种通过减少剂量来增加患者的药物依从性,并通过持续维持药物的血浆浓度来持续维持药物效果的缓释片。
[0006]
然而,传统研发的单一缓释片存在一个缺点,即需要很长时间才能在早期达到有效血浆浓度。因此,在口服给药后,很难持续24小时保持有效的药理活性,同时表现出快速的药理活性。
[0007]
因此,需要开发一种通过在短时间内达到早期有效血浆浓度,同时在维持药物的血浆浓度方面具有缓释片的优势来解决缓释片的缺点的制剂。
技术实现要素:
[0008]
技术问题
[0009]
本发明的目的是提供一种包含s(-)-西苯唑啉或其药学上可接受的盐的药物制剂,其中药物制剂包括速释层和缓释层。
[0010]
技术方案
[0011]
本发明提供了一种药物制剂,其包含约150至约450mg s(-)-西苯唑啉或其药学上可接受的盐,其中药物制剂包括速释层和缓释层。所述药物制剂可包含约150mg、约200mg、约250mg、约300mg或约400mg s(-)-西苯唑啉或其药学上可接受的盐。
[0012]
在本发明的一种实施方式中,在药物制剂中,速释层中所含的s(-)-西苯唑啉或其药学上可接受的盐与缓释层中所含的s(-)-西苯唑啉或其药学上可接受的盐的重量比可为
约1:4至约1:1。重量比可为约1:3至约1:1,约3:7至约1:1,约1:2至约1:1,约7:13至约1:1,约2:3至约1:1,约9:11至约1:1,约1:4至9:11,约1:3至约9:11,约3:7至约9:11,约7:13至约9:11,约2:3至约9:11,约1:4至2:3,约1:3至约2:3,约3:7至约2:3,约7:13至约2:3,约1:4至7:13,约1:3至约7:13,约3:7至约7:13,约1:4至3:7,约1:3至约3:7,或约1:4至约1:3。
[0013]
在本发明的一种实施方式中,药物制剂中所含的缓释剂的量可为缓释层总重量的5%至30%。药物制剂中所含的缓释剂的量可为缓释层总重量的6%至30%或7%至30%。
[0014]
在本发明的一种实施方式中,药物制剂中的缓释剂的粘度可为约1500至约200000厘泊(cps)。缓释剂的粘度可为约1550至约200000、约1600至约200000厘泊(cps)、约1500至约180000厘泊(cps)、约1550至约180000、约1600至约180000厘泊(cps)、约1500至约10000厘泊(cps)、约1550至约150000厘泊(cps),或约1600至约150000厘泊(cps)。粘度是根据美国药典(usp)推荐的测试方法测量的值。
[0015]
在本发明的一种实施方式中,缓释剂可选自羟乙基纤维素、羟丙基甲基纤维素及其盐或衍生物、聚氧化乙烯及其盐或衍生物、羧甲基纤维素钠、卡波姆、聚醋酸乙烯酯及其混合物。
[0016]
在本发明的一种实施方式中,药物制剂的速释层可含有崩解剂。
[0017]
在本发明的一种实施方式中,药物制剂中所含的崩解剂的量为速释层总重量的约1%至约10%。药物制剂中所含的崩解剂的量为缓释层总重量的约1%至约5%。
[0018]
在本发明的一种实施方式中,崩解剂可选自羧甲基纤维素钙、预糊化淀粉、淀粉乙醇酸钠、交联聚维酮(polyplasdone)、交联羧甲基纤维素钠、低取代羟丙基纤维素、海藻酸、粉状纤维素、淀粉、海藻酸钠及其混合物。
[0019]
在本发明的一种实施方式中,在整个速释层中,药物制剂的速释层可包含(a-1)s(-)-西苯唑啉或其药学上可接受的盐和(a-2)选自羧甲基纤维素钙、预糊化淀粉、淀粉乙醇酸钠、交联聚维酮(polyplasdone)、交联羧甲基纤维素钠、低取代羟丙基纤维素、海藻酸、粉状纤维素、淀粉、海藻酸钠及其混合物的崩解剂。
[0020]
在本发明的一种实施方式中,药物制剂的缓释层可包含(b-1)s(-)-西苯唑啉或其药学上可接受的盐和(b-2)选自羟乙基纤维素、羟丙基甲基纤维素及其盐或衍生物、聚氧化乙烯及其盐或衍生物、羧甲基纤维素钠、卡波姆、聚醋酸乙烯酯及其混合物的缓释剂。
[0021]
在本发明的一种实施方式中,药物制剂的速释层可包含(a-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为速释层总重量的约25%至约40%,和(a-2)选自羧甲基纤维素钙、预胶化淀粉、淀粉乙醇酸钠、交联聚维酮(polyplasdone)、交联羧甲基纤维素钠、低取代羟丙基纤维素、海藻酸、粉状纤维素、淀粉、海藻酸钠及其混合物的崩解剂,其量为速释层总重量的约1%至约10%。
[0022]
在本发明的一种实施方式中,药物制剂的缓释层可包含(b-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为缓释层总重量的约30%至约40%,和(b-2)选自羟乙基纤维素,羟丙基甲基纤维素及其盐或衍生物、聚氧化乙烯及其盐或衍生物、羧甲基纤维素钠、卡波姆、聚醋酸乙烯酯及其混合物的缓释剂,其量为缓释层总重量的约5%至约30%。
[0023]
在本发明的一种实施方式中,在整个速释层中,药物制剂的速释层可包含(a-1)s(-)-西苯唑啉或其药学上可接受的盐和(a-2)选自羧甲基纤维素钙、预糊化淀粉、淀粉乙醇酸钠、交联聚维酮(polyplasdone)、交联羧甲基纤维素钠、低取代羟丙基纤维素、海藻酸、粉
状纤维素、淀粉、海藻酸钠及其混合物的崩解剂,和
[0024]
缓释层可包含(b-1)s(-)-西苯唑啉或其药学上可接受的盐和(b-2)选自羟乙基纤维素、羟丙基甲基纤维素及其盐或衍生物、聚氧化乙烯及其盐或衍生物、羧甲基纤维素钠、卡波姆、聚醋酸乙烯酯及其混合物的缓释剂。
[0025]
在本发明的一种实施方式中,药物制剂的速释层可包含(a-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为速释层总重量的约25%至约40%,和(a-2)选自羧甲基纤维素钙、预糊化淀粉、淀粉乙醇酸钠、交联聚维酮(polyplasdone)、交联羧甲基纤维素钠、低取代羟丙基纤维素、海藻酸、粉状纤维素、淀粉、海藻酸钠及其混合物的崩解剂,其量为速释层总重量的约1%至约10%,和
[0026]
缓释层可包含(b-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为缓释层总重量的约30%至约40%,和(b-2)选自羟乙基纤维素、羟丙基甲基纤维素及其盐或衍生物、聚氧化乙烯及其盐或衍生物、羧甲基纤维素钠、卡波姆、聚醋酸乙烯酯及其混合物的缓释剂,其量为缓释层总重量的约5%至约30%。
[0027]
在本发明的一种实施方式中,药物制剂的速释层或缓释层可包含选自微晶纤维素、一水乳糖、羟丙基纤维素、硬脂酸镁、胶体二氧化硅、乳糖、糊精、甘露醇、山梨醇、微晶纤维素、磷酸氢钙、无水磷酸氢钙、碳酸钙、糖类、聚乙烯吡咯烷酮、共聚维酮(copovidone)、明胶、淀粉、蔗糖、甲基纤维素、乙基纤维素、轻质无水硅酸、滑石、硬脂酸及其混合物的赋形剂。
[0028]
在本发明的一种实施方式中,药物制剂可在溶出试验中满足双相溶出曲线。
[0029]
在本发明的一种实施方式中,药物制剂可满足以下双相溶出曲线:
[0030]
1)约30分钟内释放总重量的约30%至约50%的s(-)-西苯唑啉或其药学上可接受的盐,和
[0031]
2)此后,约12小时内实现总重量的约90%或更多的s(-)-西苯唑啉或其药学上可接受的盐的释放。
[0032]
溶出曲线还可以满足以下溶出曲线:
[0033]
1)约15分钟内释放总重量的约30%至约50%的s(-)-西苯唑啉或其药学上可接受的盐,和
[0034]
2)此后,约12小时内实现总重量的约80%或更多的s(-)-西苯唑啉或其药学上可接受的盐的释放。
[0035]
在本发明的一种实施方式中,药物制剂可满足以下溶出曲线:
[0036]
1)约30分钟内释放总重量的约30%至约50%的s(-)-西苯唑啉或其药学上可接受的盐,
[0037]
2)此后,约3小时内实现总重量的约60%或更多且约75%或更少的s(-)-西苯唑啉或其药学上可接受的盐的释放,和
[0038]
3)此后,约12小时内实现总重量的约90%或更多的s(-)-西苯唑啉或其药学上可接受的盐的释放。
[0039]
溶出曲线还可满足以下溶出曲线:
[0040]
1)约15分钟内释放总重量的约30%至约50%的s(-)-西苯唑啉或其药学上可接受的盐,
[0041]
2)此后,约3小时内实现总重量的约55%或更多且约75%或更少的s(-)-西苯唑啉或其药学上可接受的盐的释放,和
[0042]
3)此后,约12小时内实现总重量的约80%或更多的s(-)-西苯唑啉或其药学上可接受的盐的释放。
[0043]
在本发明的一种实施方式中,在药物制剂中,s(-)-西苯唑啉或其药学上可接受的盐的血浆浓度[c]在24小时的约6小时至约18小时内可为约150ng/ml或更高。此外,s(-)-西苯唑啉的血浆浓度[c]在24小时内可为约100ng/ml或更高。在本发明的一种实施方式中,即使当每天一次给药制剂时,上述时间范围内的最低血浆浓度也会出现。
[0044]
在本发明的一种实施方式中,当每天一次以200mg的单次剂量给药药物制剂时,药物制剂的cmax可为约280至约420ng/ml。
[0045]
在本发明的一种实施方式中,当每天一次以300mg的单次剂量给药药物制剂时,药物制剂的cmax可为约430至约570ng/ml。
[0046]
在本发明的一种实施方式中,当每天一次以200mg的单次剂量给药药物制剂时,药物制剂在24小时内的auc可为约1890至约2830ng
·
h/ml。
[0047]
在本发明的一种实施方式中,当每天一次以300mg的单次剂量给药药物制剂时,药物制剂在24小时内的auc可为约3390至约4330ng
·
h/ml。
[0048]
在本发明的一种实施方式中,药物制剂的速释层可含有(a-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为速释层总重量的约20%至约50%,(a-2)崩解剂,当速释层含有或不含有崩解剂时,其量为速释层总重量的约1%至约10%,(a-3)粘合剂,其量为速释层总重量的约0.5%至约5%,(a-4)润滑剂,其量为速释层总重量约0.1%至约3%,和(a-5)赋形剂,其量为速释层总重量的约40%至约60%。
[0049]
在本发明的一种实施方式中,药物制剂的缓释层可含有(b-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为缓释层总重量的约20%至约50%,(b-2)缓释剂,其量为缓释层总重量的约5%至约30%,(b-3)粘合剂,其量为缓释层总重量的约1%至约10%,(b-4)润滑剂,其量为缓释层总重量的约0.1%至约3%,和(b-5)赋形剂,其量为缓释层总重量的约为40%至约60%。
[0050]
在本发明的一种实施方式中,药物制剂的速释层可含有(a-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为速释层总重量的约30%至约50%,(a-2)崩解剂,其量为速释层总重量的约1%至约5%,(a-3)粘合剂,其量为速释层总重量的约0.5%至约2%,(a-4)润滑剂,其量为速释层总重量的约0.1%至约3%,和(a-5)赋形剂,其量为速释层总重量的约为40%至约60%。
[0051]
在本发明的一种实施方式中,药物制剂的缓释层可含有(b-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为缓释层总重量的约20%至约40%,(b-2)缓释剂,其量为缓释层总重量的约10%至约20%,(b-3)粘合剂,其量为缓释层总重量的约1%至约10%,(b-4)润滑剂,其量为缓释层总重量的约0.1%至约3%,和(b-5)赋形剂,其量为缓释层总重量的约40%至约60%。
[0052]
在本发明的一种实施方式中,药物制剂的速释层可含有(a-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为速释层总重量的约23%至约29%,可不含有(a-2)崩解剂,且可含有(a-3)粘合剂、(a-4)润滑剂和(a-5)赋形剂,其量为速释层总重量的约69%至约71%。
[0053]
在本发明的一种实施方式中,缓释层可含有(b-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为缓释层总重量的约33%至约39%,(b-2)缓释剂,其量为缓释层总重量的约9%至约10%,以及(a-3)粘合剂、(a-4)润滑剂和(a-5)赋形剂,其量为缓释层总重量的约49%至约51%。
[0054]
在本发明的一种实施方式中,药物制剂的速释层可含有(a-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为速释层总重量的约38%至约42%,(a-2)崩解剂,其量为速释层总重量的约1.2%至约5%,以及(a-3)粘合剂、(a-4)润滑剂和(a-5)赋形剂,其量为速释层总重量的约41%至约53%。
[0055]
在本发明的一种实施方式中,缓释层可含有(b-1)s(-)-西苯唑啉或其药学上可接受的盐,其量为缓释层总重量的约28%至约32%,(b-2)缓释剂,其量为缓释层总重量的约13%至约17%,以及(a-3)粘合剂、(a-4)润滑剂和(a-5)赋形剂,其量为缓释层总重量的约47%至约51%。
[0056]
在本发明的一种实施方式中,在药物制剂中,速释层中所含的崩解剂与粘合剂的重量比可为约1:2到约10:1。重量比可为约1:1至10:1、1:2至5:1或1:1至5:1。
[0057]
在本发明的一种实施方式中,在药物制剂中,缓释层中所含的缓释剂与粘合剂的重量比可为约1:60至约10:8。重量比可为约1:30至10:8,1:60至5:8,或1:30至5:8。
[0058]
在本发明的一种实施方式中,在药物制剂中,速释层中所含的(-)-西苯唑啉或其药学上可接受的盐与崩解剂的重量比可为约6:1至约50:1。重量比可为约12:1至50:1、6:1至25:1或12:1至25:1。
[0059]
在本发明的一种实施方式中,在药物制剂中,缓释层中所含的(-)-西苯唑啉或其药学上可接受的盐与缓释剂的重量比可为约1:1至约4:1。重量比可为约2:1至4:1、1:1至2:1或2:1至1:2。
[0060]
在本发明的一种实施方式中,在药物制剂中,速释层中所含的(-)-西苯唑啉或其药学上可接受的盐与粘合剂的重量比可为约15:1至约100:1。重量比可为约30:1至100:1、15:1至50:1或30:1至50:1。
[0061]
在本发明的一种实施方式中,在药物制剂中,缓释层中所含的(-)-西苯唑啉或其药学上可接受的盐与粘合剂的重量比可为约2:1至约40:1。重量比可为约4:1至40:1、2:1至20:1或4:1至20:1。
[0062]
本发明提供了一种包括药物制剂的用于治疗肥厚型心肌病的药物制剂。
[0063]
本发明提供了一种试剂盒,包括药物制剂和指示患者每天一次给药药物制剂的说明书。
[0064]
有益效果
[0065]
根据本发明的包括速释层和缓释层的药物制剂具有双相溶出曲线,因此可以在早期快速达到s(-)-西苯唑啉的有效血浆浓度,并且可以在后期持续保持有效血浆浓度。因此,s(-)-西苯唑啉的药效可以仅通过每天一次单次剂量给药来维持。
附图说明
[0066]
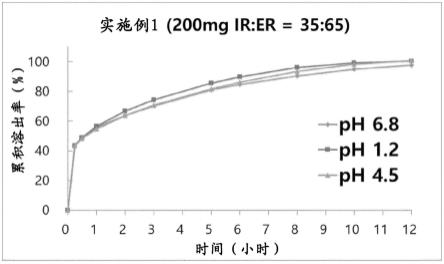
图1是示出了本发明实施例1的缓释制剂在ph 6.8、ph 1.2和ph 4.5下的溶出速率的图。
[0067]
图2是示出了本发明实施例2的缓释制剂在ph 6.8、ph 1.2和ph 4.5下的溶出速率的图。
[0068]
图3是示出了本发明实施例3的缓释制剂在ph 6.8、ph 1.2和ph 4.5下的溶出速率的图。
具体实施方式
[0069]
为了更容易理解本发明,本发明中使用的术语定义如下。
[0070]
本发明中术语“约”旨在包括所示出的本发明的目的和效果的范围。例如,在所含的缓释剂的量为缓释层总重量的约5%至30%的情况下,可将其解释为可延伸至本领域技术人员通过已知技术示出的本发明的目的和效果的范围以及5%至30%的范围。
[0071]
西苯唑啉或其药学上可接受的盐
[0072][0073]
西苯唑啉(cifenline,2-(2,2-二苯基环丙基)-4,5-二氢-1h-咪唑,53267-01-9(cas号))是一种具有化学式1结构的化合物,并且是一种用作抗心律失常剂的药物(1族钠通道阻断剂)。此外,西苯唑啉作为钙通道阻滞剂和钾通道阻滞剂的作用也是已知的。
[0074]
西苯唑啉作为两种对映体存在,例如s(-)-西苯唑啉和r(+)-西苯唑啉,目前市场上可买到的西苯唑啉是一种s(-)-西苯唑啉和r(+)-西苯唑啉以1:1的比例(外消旋混合物:50:50的对映体混合物)混合的组合物。在本发明中,s(-)-西苯唑啉用作西苯唑啉。
[0075]
本发明的s(-)-西苯唑啉不仅可以含有西苯唑啉的游离盐,而且还可以含有其氯化物,并且西苯唑啉不仅可以是药学上可接受的盐或所有水合物和溶剂化物。在一种实施方式中,水合物或溶剂化物可为在将西苯唑啉溶解于水溶性溶剂(例如甲醇、乙醇、丙酮或1,4-二氧六环)中,然后添加游离酸或游离碱后获得的结晶或再结晶的溶剂化物(尤其是水合物)。此外,还可包括化学计量溶剂化物,其包括可通过诸如冻干之类的方法制备的各种量的含水水合物。
[0076]
西苯唑啉的氯化物包括无机酸盐和有机酸盐,其实例包括但不限于盐酸盐、硫酸盐、硝酸盐、磷酸盐、醋酸盐、三氟乙酸盐、苯磺酸盐、琥珀酸盐、酒石酸盐、马来酸盐、柠檬酸盐等。
[0077]
西苯唑啉可配制以给药于患者,并含有一种或多种生理上可接受的载体或赋形剂。该载体可与制剂的其他组分相容且对接受者无害,并且该药物可配制成口服、静脉注射或直肠给药。作为该制剂的实例,该药物可被配制成一般形式,例如片剂、胶囊和糖浆,但本发明不限于此。
[0078]
剂量
[0079]
本发明的药物制剂的合适剂量可通过多种方式选择,具体取决于配制方法、给药方式和患者的年龄、体重、性别、病理状态、食物、给药时间、给药途径、排泄率和反应敏感性等因素。
[0080]
在本发明的一种实施方式中,本发明的药物制剂的每天一次剂量可为约150mg至约450mg、约150mg至约400mg或约200mg至约400mg,但不限于此。
[0081]
此外,在本发明的一种实施方式中,本发明的药物制剂的每天一次剂量可为150mg、160mg、170mg、180mg、190mg、200mg、210mg、220mg、230mg、240mg、250mg、260mg、270mg、280mg、290mg、300mg、310mg、320mg、330mg、340mg、350mg、360mg、370mg、380mg、390mg、400mg、410mg、420mg、430mg、440mg,或450mg,但不限于此。
[0082]
药物制剂
[0083]
本发明的药物制剂包括速释层(ir层)和缓释层(er层)。速释层指的是其中s(-)-西苯唑啉或其药学上可接受的盐相对于缓释层首先被释放到活体的制剂,但不限于此配置。
[0084]
在本发明的一种实施方式中,速释层和缓释层中的每一种的制剂可以是多颗粒制剂、药物制剂、球形制剂或双层(双层)片剂,但不限于此。
[0085]
在本发明的一种实施方式中,药物制剂可包括含有s(-)-西苯唑啉或其药学上可接受的盐的颗粒、含有西苯唑啉或其药学上可接受的盐的药物包衣球体,或至少两个含有西苯唑啉或其药学上可接受的盐的层群。药物制剂的第一层群立即在上消化道释放药物,并且药物制剂的第二层群在消化道的下部释放药物。在本发明的一种实施方式中,第二层群可被ph依赖性涂层或时间依赖性涂层包覆,以延迟药物第二次释放到胃肠道中的所需位置。
[0086]
在本发明的一种实施方式中,制剂的速释层中所含的s(-)-西苯唑啉或其药学上可接受的盐:缓释层中所含的西苯唑啉或其药学上可接受的盐的重量比可为约1:4至约1:1、约1:3至约1:1、约3:7至约1:1、约1:2至约1:1、约7:13至约1:1、约2:3至约1:1、约9:11至约1:1、约1:4至9:11、约1:3至约9:11、约3:7至约9:11、约7:13至约9:11、约2:3至约9:11、约1:4至2:3、约1:3至约2:3、约3:7至约2:3、约7:13至约2:3、约1:4至7:13、约1:3至约7:13、约3:7至约7:13、约1:4至3:7、约1:3至约3:7或约1:4至约1:3。
[0087]
当速释层中所含的s(-)-西苯唑啉或其药学上可接受的盐:缓释层中所含的西苯唑啉或其药学上可接受的盐的重量比小于约1:4时,给药后很难立即获得有效效果,并且当速释层中所含的s(-)-西苯唑啉或其药学上可接受的盐:缓释层中所含的西苯唑啉或其药学上可接受的盐的重量比超过约1:1时,由于其在缓释层中的量减少,因此难以获得适当的缓释效果。
[0088]
缓释层
[0089]
在本发明的一种实施方式中,缓释层含有缓释剂,以便具有s(-)-西苯唑啉或其药学上可接受的盐的缓释效果。
[0090]
在本发明的一种实施方式中,缓释剂用于在达到治疗性血浆浓度后持续维持药物一段时间,同时比普通制剂中的药物缓慢释放更长的时间。
[0091]
在本发明的一种实施方式中,缓释剂的粘度可为约1500至约200000厘泊(cps)、约
1550至约200000厘泊(cps)、约1600至约200000厘泊(cps)、约1500至约180000厘泊(cps)、约1550至约180000厘泊(cps)、约1600至约180000厘泊(cps)、约1500至约10000厘泊(cps)、约1550到150000,或约1600到150000厘泊(cps)。当粘度低于约1500厘泊或高于约200000厘泊时,根据s(-)-西苯唑啉或其药学上可接受的盐的药代动力学行为,可能难以实现优选溶出模式。
[0092]
在本发明的一种实施方式中,作为缓释剂,使用选自离子剂、溶胀剂和疏水剂中的一种或两种以上的混合物。
[0093]
在本发明的一种实施方式中,离子剂是指通过药物的离子键控制药物释放的药学上可接受的试剂,并包括选自羧甲基纤维素钠、卡波姆和海藻酸钠中的一种或多种。
[0094]
在本发明的一种实施方式中,溶胀剂是指在水溶液中瞬间溶胀并通过减少孔隙控制药物释放的药学上可接受的试剂,包括选自羟乙基纤维素及其盐或衍生物、羟丙基甲基纤维素及其盐或衍生物、聚氧化乙烯及其盐或衍生物和卡拉胶中的一种或多种。
[0095]
在本发明的一种实施方式中,疏水剂是指不溶于水溶液并通过孔阻塞控制药物释放的药学上可接受的试剂,包括选自聚醋酸乙烯酯、苯甲酸甘油酯、氢化蓖麻油、氢化植物油和硬脂酸中的一种或多种。
[0096]
在本发明的一种实施方式中,缓释剂包括选自羟乙基纤维素、羟丙基甲基纤维素及其盐或衍生物、聚氧化乙烯及其盐或衍生物、羧甲基纤维素钠、卡波姆和聚醋酸乙烯酯中的一种或多种。
[0097]
在本发明的一种实施方式中,相对于100重量份的缓释层,缓释剂的量为约5至约30重量份、约6至约30重量份或约7至约30重量份。当缓释剂的量为约5至约30重量份时,药物释放控制可能是极好的,并且可以以适当的西苯唑啉或其药学上可接受的盐的释放速率达到最佳血浆浓度。当缓释剂的量超过约30重量份时,药物在早期的释放缓慢,约12小时内100%的药物未溶解,药物的大小增加。当缓释剂的量小于约5重量份时,药物在早期的释放很快,并且约12小时内整个药物提前释放。
[0098]
速释层
[0099]
在本发明的一种实施方式中,速释层是含有崩解剂的制剂或不含缓释剂的制剂,以便具有s(-)-西苯唑啉或其药学上可接受的盐的速释效果。
[0100]
在本发明的一种实施方式中,崩解剂用于吸收水分并促进制剂的崩解,以改善溶解。
[0101]
在本发明的一种实施方式中,崩解剂包括选自羧甲基纤维素钙、预糊化淀粉、淀粉乙醇酸钠、交联聚维酮(polyplasdone)、交联羧甲基纤维素钠、低取代羟丙基纤维素、海藻酸、粉状纤维素、淀粉、海藻酸钠及其混合物中的一种或多种。
[0102]
在本发明的一种实施方式中,相对于100重量份的速释层,崩解剂的量为约1至约10重量份或约1至约5重量份。当崩解剂的含量太低时,溶解没有改善。此外,当崩解剂的含量过多时,最终制剂的硬度会受到不利影响,并且制剂容易破碎。
[0103]
赋形剂
[0104]
在本发明的一种实施方式中,为了确保长期储存的稳定性,并具有持续的缓释效果,可额外含有赋形剂。在本发明中,赋形剂指稀释剂、粘合剂、润滑剂等。
[0105]
例如,在本发明的一种实施方式中,稀释剂可选自由乳糖、一水乳糖、糊精、甘露
醇、山梨醇、淀粉、微晶纤维素、磷酸氢钙、无水磷酸氢钙、碳酸钙、糖类及其混合物组成的组。
[0106]
在本发明的一种实施方式中,粘合剂用于增加制剂的结合力。粘合剂可选自由聚乙烯吡咯烷酮、共聚维酮、明胶、蔗糖、甲基纤维素、乙基纤维素及其混合物组成的组。
[0107]
在本发明的一种实施方式中,润滑剂改善了粉末和颗粒材料的流动性,以增加对位于压片机下部的模具的填充能力,并减少粉末和颗粒材料之间以及粉末和颗粒材料与位于压片机上部的冲头和模具之间的摩擦,从而促进片剂的压缩和释放。例如,润滑剂可以是轻质无水硅酸、胶体二氧化硅、滑石、硬脂酸、硬脂酸镁及其混合物。
[0108]
溶出试验
[0109]
溶出试验是根据美国药典(或韩国药典)中溶出方法ii(桨叶法)进行的试验。
[0110]
双相曲线
[0111]
在本发明的一种实施方式中,制剂的双相溶出曲线可包括作为第一阶段的速释阶段(或瞬时释放阶段)和第二阶段的缓释阶段(或延迟释放阶段)。
[0112]
第一阶段是在体外溶出试验中的0至约30分钟内获得的溶出曲线的一部分,并且总重量的30至50%的s(-)-西苯唑啉或其药学上可接受的盐在第一相开始的约30分钟内释放。“约30分钟内”指给药后经过一定时间的时间点,包括约10分钟内、约15分钟内或约20分钟内。
[0113]
第二阶段是在体外溶出试验中测量的约30分钟后的溶出曲线的一部分。“约30分钟后”指给药后经过一定时间的时间点,包括约10分钟后、约15分钟后或约20分钟后。
[0114]
在本发明的一种实施方式中,在制剂中,总重量的约50%至约90%或约50%至80%的s(-)-西苯唑啉或其药学上可接受的盐从第二阶段中释放。
[0115]“cmax”、“cmin”、“tmax”、“tmin”、“auc”[0116]
本发明中的“cmax”、“cmin”、“tmax”和“tmin”是用于药物浓度随时间变化的药代动力学分析的术语。cmax是表示在给药后和第二药物剂量给药前,药物在身体特定隔间或测试区域内达到的最大(或峰值)血浆浓度的术语。cmax是与cmin相反的术语,cmin是给药后药物达到的最低(或最低)浓度。tmax是药代动力学中用于描述观察到cmax的时间的术语,tmin是药代动力学中用于描述给药后和第二药物剂量给药前观察到cmin的时间的术语。
[0117]
在本发明的一种实施方式中,当每天一次以200mg的单次剂量给药制剂时,该制剂的cmax为约280至约420ng/ml。此外,当每天一次以300mg的单次剂量给药制剂时,该制剂的cmax为约430至约570ng/ml。至于cmax值,代表药效学分析效果的适当数值可能会随着剂量和给药次数的增加而增加。在本发明的一种实施方式中,制剂的cmax在给药制剂后满足约1至约3小时(即tmax)。
[0118]
本发明中的“auc”是指药物浓度随时间变化的药代动力学图中曲线下的面积(数学上称为定积分)。
[0119]
在本发明的一种实施方式中,当每天一次以200mg的单次剂量给药制剂时,该制剂在24小时内满足约1890至约2830ng
·
h/ml的目标曲线下面积(以下称为曲线下面积(auc))。此外,当每天一次以300mg的单次剂量给药制剂时,该制剂的auc为约3390至约4330ng
·
h/ml。至于auc值,代表药效学分析效果的适当数值可能会随着剂量和给药次数的
增加而增加。
[0120]
在本发明的一种实施方式中,在24小时的约6小时至约18小时内,该制剂中的s(-)-西苯唑啉的血浆浓度[c]约为150ng/ml或更高。此外,24小时内的s(-)-西苯唑啉的血浆浓度[c]约为100ng/ml或更高。在本发明的一种实施方式中,即使每天一次给药制剂,上述时间范围内的最低血浆浓度也会出现。
[0121]
药物组合物
[0122]
在向患者给药本发明的药物制剂后约0.25至约1小时内达到最低有效血浆浓度。在本发明的一种实施方式中,在药物制剂的速释层中实现约150至约800ng/ml的最小有效血浆浓度。
[0123]
肥厚型心肌病(hcm)
[0124]
肥厚型心肌病包括一组高表达的单基因常染色体显性心肌疾病,由构成心肌功能单位肌节的任何一个结构蛋白基因中1000多个已知点突变中的一个或多个引起。肥厚型心肌病最初被认为是一种非常罕见的疾病。然而,目前已经发现,肥厚型心肌病是最常见的疾病,其发病率高达每500例新生儿中就有1例,并导致猝死,尤其是在年轻时,并且以常染色体显性方式遗传。肥厚型心肌病通常在年轻人进行心电图和超声心动图健康检查时偶然发现,然后进行随访。
[0125]
血流动力学或症状性肥厚型心肌病可分为梗阻型和非梗阻型。梗阻型可分为瓣膜下梗阻和心室中心性梗阻,延迟梗阻型是指在静止时没有压力梯度,但在激发期间具有30mmhg或更大的压力梯度的情况。
[0126]
肥厚型心肌病患者的临床症状是劳累时呼吸困难,包括全身动脉血栓栓塞疾病,如中风、急性肺水肿、心房颤动、低血容量或高血容量不耐受和晕厥。最近,心脏性猝死(scd)也被确定为肥厚型心肌病的主要症状。
[0127]
试剂盒
[0128]
试剂盒是指含有用于给药本发明的s(-)-西苯唑啉或其药学上可接受的盐以治疗肥厚型心肌病的组分的包装产品。该试剂盒包括容器或盒子,用于存放试剂盒的组分。盒子或容器附有食品和药物管理局批准的方案或标签。盒子或容器保留本发明包含在塑料、聚乙烯、聚丙烯、乙烯或丙烯容器中的组分。容器可以是带盖的管子或瓶子。试剂盒还包括用于给药本发明的s(-)-西苯唑啉或其药学上可接受的盐的说明书。
[0129]
下文将参考以下实施例详细描述本发明。然而,以下实施例仅为描述本发明的实施例,本发明的范围不限于此。
[0130]
实施例1至3:根据s(-)-琥珀酸西苯唑啉的含量制备药物制剂
[0131]
根据表1的成分和含量所示的s(-)-琥珀酸西苯唑啉的含量制备双层片剂。s(-)-琥珀酸西苯唑啉的总含量分别设置为实施例1中200mg、实施例2中300mg和实施例3中400mg。详细的制备过程如下。
[0132]
[实施例1和2的制备过程]
[0133]
《步骤1》速释层的制备
[0134]
将羟丙基纤维素在室温下溶解在纯化水中,以制备结合溶液。通过30目筛过筛的s(-)-琥珀酸西苯唑啉、微晶纤维素和一水乳糖在高速混合器(hsm)(pilotmix 150t,huttlin)中混合约5分钟。将制备的结合溶液添加到混合物中,并在高速混合器中制备颗粒
约5分钟。对制备的湿颗粒进行湿磨,然后将湿磨颗粒移至流化床造粒机(fbg)(pilotlab l,huttlin)中进行干燥。将干燥后的颗粒通过锥形磨机进行大小分级。将通过30目筛过筛的羧甲基纤维素钙和微晶纤维素与大小分级的颗粒混合约22分钟。将通过40目筛过筛的硬脂酸镁在制备的混合物中润滑约5分钟,制备速释层。
[0135]
《步骤2》缓释层的制备
[0136]
将羟丙基纤维素在室温下溶解在纯化水中,以制备结合溶液。通过30目筛过筛的s(-)-琥珀酸西苯唑啉、微晶纤维素和一水乳糖在高速混合器(hsm)(pilotmix 150t,huttlin)中混合约5分钟。将制备的结合溶液添加到混合物中,并在高速混合器中制备颗粒约5分钟。对制备的湿颗粒进行湿磨,然后将湿磨颗粒移至流化床造粒机(fbg)(pilotlab l,huttlin)中进行干燥。干燥后的颗粒通过锥形磨机进行大小分级。将通过30目筛过筛的羟丙基甲基纤维素、羟丙基纤维素和微晶纤维素与大小分级的颗粒混合约17分钟。将通过40目筛过筛的硬脂酸镁在制备的混合物中润滑约5分钟,制备缓释层。
[0137]
《步骤3》双层片剂的制备
[0138]
使用双层压片机(双层压片机,kilian)将制备的速释层和缓释层颗粒制备成包括各层的双层片剂。使用制备的欧巴代(opadry)进行薄膜包衣。
[0139]
[实施例3的制备过程]
[0140]
《步骤1》速释层的制备
[0141]
将通过30目筛过筛的s(-)-琥珀酸西苯唑啉、微晶纤维素、羟丙基纤维素和一水乳糖在室温下在高速混合器(hsm)(pilotmix 150t,huttlin)中混合约10分钟。向混合物中加入纯化水,并在高速混合器中混合约5分钟,从而制备颗粒。对制备的湿颗粒进行湿磨,然后将湿磨颗粒移至流化床造粒机(fbg)(pilotlab l,huttlin)中进行干燥。将干燥后的颗粒通过锥形磨机进行大小分级。将通过30目筛过筛的胶体二氧化硅与大小分级的颗粒混合约17分钟。将通过40目筛过筛的硬脂酸镁在制备的混合物中润滑约5分钟,制备速释层。
[0142]
《步骤2》缓释层的制备
[0143]
将通过30目筛过筛的s(-)-琥珀酸西苯唑啉、微晶纤维素、羟丙基纤维素和一水乳糖在室温下在高速混合器(hsm)(pilotmix 150t,huttlin)中混合约10分钟。向混合物中加入纯化水,并在高速混合器中混合约5分钟,从而制备颗粒。对制备的湿颗粒进行湿磨,然后将湿磨颗粒移至流化床造粒机(fbg)(pilotlab l,huttlin)中进行干燥。将干燥后的颗粒通过锥形磨机进行大小分级。将通过30目筛过筛的羟丙基甲基纤维素、羟丙基纤维素和胶体二氧化硅与大小分级的颗粒混合约17分钟。将通过40目筛过筛的硬脂酸镁在制备的混合物中润滑约5分钟,制备缓释层。
[0144]
《步骤3》双层片剂的制备
[0145]
使用双层压片机(双层压片机,kilian)将制备的速释层和缓释层颗粒制备成包括各层的双层片剂。使用制备的欧巴代(opadry)进行薄膜包衣。
[0146]
[表1]
[0147][0148][0149]
表中的含量(w%)为重量份。
[0150]
实施例4至7:根据缓释层中缓释剂的含量(w%)制备制剂
[0151]
为了确认表1中实施例1的缓释层组合物中缓释剂含量的影响,通过调整作为缓释剂的羟丙基甲基纤维素的含量制备实施例4至6的制剂。通过调整作为实施例3的缓释层组合物中的缓释剂的羟丙基甲基纤维素的含量来制备实施例6至9的制剂。缓释剂的调整重量和缓释层中缓释剂的调整含量(w%)如表2所示。
[0152]
[表2]
[0153][0154]
实施例10至15和对比例1至4:根据速释层中所含的s(-)-琥珀酸西苯唑啉与缓释层中所含的s(-)-琥珀酸西苯唑啉的重量比制备制剂
[0155]
基于表1中所示的实施例1的组成,为了确认根据速释层和缓释层中所含的s(-)-琥珀酸西苯唑啉之间比例的效果,通过改变速释层中所含的s(-)-琥珀酸西苯唑啉和缓释层中所含的s(-)-琥珀酸西苯唑啉的重量比制备实施例10至15和对比例1至4的制剂。改变后的比例如表3所示。
[0156]
[表3]
[0157]
[0158][0159]
实施例16和17:根据羟丙基甲基纤维素的粘度制备制剂
[0160]
基于表1中所示的实施例1的组成,通过将作为缓释层中所含的缓释剂的羟丙基甲基纤维素的粘度从2700cps改变至5040cps来制备实施例16和17的制剂。改变后的粘度如表4所示。
[0161]
[表4]
[0162][0163]
实验例1:体外溶出试验[溶出试验]
[0164]
根据美国药典(usp)测试方法(见《711》溶出度,仪器2)使用溶出装置(evo6300,distek)进行溶出试验,如下所示。
[0165]
《溶出试验条件》
[0166]
作为溶出溶液,使用以下四种溶液中的一种或多种。
[0167]-1)磷酸盐缓冲溶液900ml(ph 6.8)
[0168]-2)0.1n hcl水溶液900ml(ph值1.2)
[0169]-3)醋酸盐缓冲溶液900ml(ph值4.5)
[0170]-4)蒸馏水900ml
[0171]
溶出温度:37℃
[0172]
桨叶转速:50rpm
[0173]
《高效液相色谱(hplc)条件》
[0174]
色谱柱:inertsil ods-3v(4.6
×
150mm,5μm)
[0175]
流动相:磷酸盐缓冲溶液(含1-辛烷磺酸钠):乙腈=650:350(v/v)
[0176]
检测器:紫外线(uv)检测器
[0177]
流速:1.5ml/min
[0178]
注射量:5μl
[0179]
在溶出试验条件下,在15分钟、30分钟、60分钟、120分钟、180分钟、300分钟、360分钟、480分钟、600分钟和720分钟获得溶出溶液,并过滤获得的溶出溶液以获得试验溶液。
[0180]
此外,精确称量55.5mg s(-)-琥珀酸西苯唑啉,并将其置于50ml烧瓶中,用溶出溶液溶解s(-)-琥珀酸西苯唑啉,然后进行标记。根据测试溶液浓度适当稀释该溶液,并用作标准溶液。此外,在hplc分析条件下分析样品溶液和标准溶液。
[0181]
实验例1-a:根据缓释层中缓释剂的含量(w%)对实施例1至9进行溶出试验
[0182]
根据表2所示的缓释层中缓释剂的含量(w%)进行溶出试验后,在所有实施例1至9中,累积溶出量在约30分钟内为约30%至约50%,在约3小时内为约60%或更多且约75%或更少,在约12小时内为约90%或更多。此外,累积溶出量在约15分钟内为约30%至约50%,在约3小时内为约55%或更多且约75%或更少,在约12小时内约为80%或更多。
[0183]
实验例1-b:根据速释层中所含的s(-)-琥珀酸西苯唑啉和缓释层中所含的s(-)-琥珀酸西苯唑啉的重量比对实施例11至17和对比例1至4进行溶出试验
[0184]
如表5所示,表3和4的实施例11至17的累积溶出量在约30分钟内为约30%至约50%,在约3小时内为约60%或更多且约75%或更少,在约12小时内为约90%或更多,且累积溶出量在约15分钟内为约30%至约为50%,在约3小时内为约55%或更多且约75%或更少,在约12小时内为约80%或更多。另一方面,对比例1的制剂为单一缓释制剂,而对比例2的制剂为缓释层中所含的s(-)-琥珀酸西苯唑啉的含量为整个片剂的约90%(1/9的比例)的制剂,并且在这些制剂的情况下,累积溶出量在约30分钟内为小于约30%,因此,初次给药后难以发挥该制剂的药用效果。对比例3的制剂是缓释层中所含的s(-)-琥珀酸西苯唑啉的含量为整个片剂的约45%(9/20的比例)的制剂,对比例4的制剂为单一速释制剂,在这些制剂的情况下,累积溶出量在约6小时内超过约80%,因此难以获得缓释效果。
[0185]
[表5]
[0186][0187]
实验例1-c:根据ph变化的s(-)-琥珀酸西苯唑啉缓释制剂的累积溶出量(%)
[0188]
如表6和图1至3中所示,确认了根据活体消化系统的ph环境,实施例1至3的累积溶出量在约30分钟内为约30%至约50%,在约3小时内为约60%或更多且约75%或更少,在约12小时内为约90%或更多,累积溶出量在约15分钟内为约30%至50%,在约3小时内为约55%或更多且约75%或更少,在约12小时内为约80%或更多。此外,如图1至3所示,确认了在早期阶段,溶出曲线斜率在1小时之前和1小时之后发生变化的双相溶出曲线满足要求。
[0189]
[表6]
[0190][0191]
“‑”
由于测量误差,该值未得到确认
[0192]
实验例2:体内临床前试验
[0193]
在实验例1的体外溶出试验中,通过以下临床前试验确认了实施例1的缓释制剂的体内药代动力学,在该缓释制剂中,不同ph条件下的溶出方面得到了确认。
[0194]
对于试验方法,将不包括作为实施例1的缓释剂的羟丙基甲基纤维素的制剂和对比例5的制剂的各片剂口服给药于比格犬(雄性,n=6),在预定时间采集血液,分离血浆,然后测量比格犬血浆中s(-)-琥珀酸西苯唑啉的浓度。给药后每0.33、0.66、1、1.5、2、2.5、3、4、6、8、10、12、16和24小时收集1.5ml血液。通过lc-ms/ms分析方法分析血液中s(-)-琥珀酸西苯唑啉的含量,并使用phoenixent-version 8.1程序分析药代动力学参数。
[0195]
达到最大血浆浓度的时间(tmax)、最大血浆浓度(cmax)、高达24小时定量时间的血浆浓度-时间曲线下面积(auc0-24小时)、从给药开始至无限时间的血浆浓度-时间曲线下面积(auc0-inf),并计算表7所示试验的分析结果的血浆半衰期(t1/2)。
[0196]
[表7]
[0197]
药代动力学参数实施例1对比例5t
max
(h)2.0(0.66-3.0)0.66(0.33-1.50)c
max
(ng/ml)2110.0473160.389auc
0-24h
(ng
·
h/ml)14179.93213987.574auc
0-inf
(ng
·
h/ml)14279.12814094.982t
1/2
(h)2.9283.132
[0198]
如表7所示,确认在实施例1中,cmax值相对于对比例5降低了约1.5倍,tmax值增加了约3倍,并且获得了缓释效果。通过本实验可以确认,达到最大血浆浓度的时间(tmax)被延迟,因此,体外溶出方面和体内溶出方面彼此相同。基于该实验,在临床试验中确认最佳效果是本发明的最终目标。
[0199]
实验例3:在实施例1和实施例2(单次给药)的健康受试者中确认的药效学分析
[0200]
本研究是一项通过向健康受试者每天一次口服给药200mg或300mg具有实施例1和2各自组成的s(-)-琥珀酸西苯唑啉缓释片制剂,或每天三次口服给药150mg具有对比例5组成的琥珀酸西苯唑啉外消旋混合物(s-异构体:r-异构体=50:50)以评估一天的安全性和耐受性的临床试验。更具体地说,临床试验通过如下给药方式进行。
[0201]-队列1:每天一次单次口服给药200mg s(-)-琥珀酸西苯唑啉
[0202]-队列2:每日一次单次口服给药300mg s(-)-琥珀酸西苯唑啉
[0203]-队列3:每天三次单次口服给药150mg琥珀酸西苯唑啉外消旋混合物(s-异构体:r-异构体=50:50)
[0204]
在本研究中,共有24名受试者以1:1:1的比例随机分配到三个队列,并进行了筛查、治疗期1、停药期和治疗期2四个阶段。给药组与未给药组(安慰剂)的比例为6:2。
[0205]
作为主要评估变量,测量了血浆浓度-时间曲线下面积(auc)、药物的最大血浆浓度(cmax)、达到最大血浆浓度的时间(tmax)和最大血浆浓度的半衰期(t1/2),以便对健康受试者的s(-)-琥珀酸西苯唑啉的剂量增加进行血液药代动力学分析。
[0206]
当参与队列1和队列2时,在48小时内(服用试验药物前60分钟内,以及服用试验药物后每30分钟、1小时、1小时30分钟、2小时、2小时30分钟、3小时、4小时、6小时、8小时、10小时、12小时、16小时、24小时、30小时、36小时和48小时)进行药代动力学血液采集。当参与队列3时基于,在服用ct-g11前的上午8点给药、以及服用ct-g11之后(在下午4点服用ct-g11作为第二剂之前进行)的每30分钟、1小时、1小时30分钟、2小时、2小时30分钟、3小时、4小时和8小时进行药代动力学血液采集,以及在第二次给药试验药物后(在第三次给药试验药物前进行)的30分钟、1小时、1小时30分钟、2小时、2小时30分钟、3小时、4小时和8小时进行药代动力学血液采集。在第三次给药试验药物后的30分钟、1小时、1小时30分钟、2小时、2小时30分钟、3小时和4小时后分别进行采血。
[0207]
作为s(-)-琥珀酸西苯唑啉的血浆浓度,使用lc-ms从预处理的血样中测量s(-)-琥珀酸西苯唑啉的血浆浓度。如表8所示,已确认三种制剂显示出发挥药效等效的药物作用,并且最低有效血浆浓度保持在约100至150ng/ml或更高,最低有效血浆浓度通过之前进行的比格犬非临床(体内)实验得到确认,因此,药物在患者体内的作用维持了24小时。
[0208]
[表8]
[0209][0210]
实验例4:在实施例1和实施例2(多次给药)的健康受试者中确认的药效学分析
[0211]
本研究是一项通过向健康受试者每天一次口服给药200mg或300mg具有实施例1和2各自组成的s(-)-琥珀酸西苯唑啉缓释片制剂,或每天三次口服给药150mg具有对比例5组成的琥珀酸西苯唑啉外消旋混合物(s-异构体:r-异构体=50:50)以评估安全性和耐受性的临床试验。更具体地说,临床试验通过如下给药方式进行。
[0212]-队列4:每天一次,连续5天多次口服给药200mg s(-)-琥珀酸西苯唑啉
[0213]-队列5:每天一次,连续14天多次口服给药300mg s(-)-琥珀酸西苯唑啉
[0214]-队列6:每天三次,连续14天多次口服给药150mg琥珀酸西苯唑啉外消旋混合物(s-异构体:r-异构体=50:50)
[0215]
在本研究中,共有24名受试者以1:1:1的比例随机分配到三个队列,并进行了筛查、治疗期1、停药期和治疗期2四个阶段。给药组与未给药组(安慰剂)的比例为6:2。
[0216]
作为主要评估变量,测量了血浆浓度-时间曲线下面积(auc)、药物的最大血浆浓度(cmax)、达到最大血浆浓度的时间(tmax)和最大血浆浓度的半衰期(t1/2),以便对健康受试者的s(-)-琥珀酸西苯唑啉的剂量增加进行血液药代动力学分析。
[0217]
当参与队列4时,进行药代动力学血液采集6天(第3天和第4天服用试验药物前60分钟内,第14天服用试验药物前60分钟内,服用试验药物后每30分钟、1小时、1小时30分钟、2小时、2小时30分钟、3小时、4小时、6小时、8小时、10小时、12小时和16小时,以及第6天服用试验药物后24小时)。当参与队列5时,进行药代动力学血液采集15天(第3天、第4天和第5天服用试验药物前60分钟内,第14天服用试验药物前60分钟内,服用试验药物后每30分钟、1小时、1小时30分钟、2小时、2小时30分钟、3小时、4小时、6小时、8小时、10小时、12小时和16小时,以及第15天服用药物后24小时)。当参与队列6时,基于14天的上午8点给药,在第3天、第4天和第5天服用ct-g11前60分钟内进行药代动力学血液采集,以及在第14天服用ct-g11前60分钟内和给药后的每30分钟、1小时、1小时30分钟、2小时、2小时30分钟、3小时、4小时和8小时进行药代动力学血液采集。
[0218]
作为s(-)-琥珀酸西苯唑啉的血浆浓度,使用lc-ms从预处理的血液样本中测量s(-)-琥珀酸西苯唑啉的血浆浓度。如表9所示,已确认三种制剂显示出发挥药效等效的药物作用,并且最低有效血浆浓度保持在约100至150ng/ml或更高,最低有效血浆浓度通过之前进行的比格犬非临床(体内)实验得到确认,因此,药物在患者体内的作用维持了24小时。
[0219]
[表9]
[0220]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1