可用作IGE调节剂的四氢苯并-喹啉磺酰胺衍生物的制作方法
可用作ige调节剂的四氢苯并-喹啉磺酰胺衍生物
技术领域
1.本发明涉及式(i)的四氢苯并-异喹啉磺酰胺衍生物、制备它们的方法、包含它们的药物组合物以及它们在治疗由ige引起的病症(例如过敏反应、非变应性肥大细胞反应或某些自身免疫应答)且特别是由ige与fcεri受体相互作用引起的病症中的用途。
2.发明背景
3.ige(免疫球蛋白e)为免疫球蛋白家族的成员,并且介导过敏反应,例如哮喘、食物过敏、1型超敏反应和常见的窦炎。
4.ige由b细胞分泌并在b细胞的表面上表达。由b细胞合成的ige通过跨膜结构域锚定在b细胞膜中,所述跨膜结构域通过短膜结合区与成熟ige序列连接。ige还通过其fc区至低亲和力ige受体(fcεrii)而结合至b细胞(和单核细胞、嗜酸性粒细胞和血小板)。当哺乳动物暴露于变应原时,b细胞克隆扩增,其合成结合变应原的ige。该ige又被b细胞释放到循环中,在循环中ige被b细胞(通过fcεrii)和肥大细胞和嗜碱性粒细胞通过在肥大细胞和嗜碱性粒细胞表面上发现的所谓的高亲和力受体(fcεri)结合。这样的肥大细胞和嗜碱性粒细胞由此对变应原致敏。下次暴露于变应原使这些细胞上的fcεri交联,从而激活它们释放组胺和引起临床超敏反应和过敏反应的其它因子。
5.目前,过敏性疾病、荨麻疹和哮喘通常用以下药物的一种或多种治疗:(1)拮抗炎性介体组胺和白三烯的抗组胺药和抗白三烯药,(2)抑制广谱炎性机制的局部或系统(口服或注射)皮质类固醇或免疫抑制剂,(3)短效或长效支气管扩张剂,其松弛哮喘中收缩气道的平滑肌,或(4)肥大细胞稳定剂,其抑制通常由fcεri上的ige结合触发的肥大细胞脱粒,(5)防止ige在fcεri上结合的生物制品。也曾一直尝试使用调节ige与fcεri结合的肽。例如,wo96/01643描述了治疗即时过敏反应的由4-50个氨基组成的肽。
6.但是,仍然需要鉴定在治疗或预防由ige引起的病症,特别是由ige与fcεri受体相互作用引起的病症中具有治疗效用的化合物。
7.发明概述已发现式(i)的化合物及其药学上可接受的盐可以用于该目的。
8.详细描述
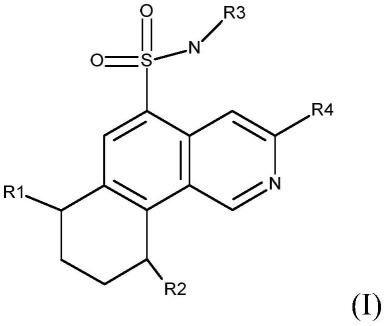
9.本发明提供了式(i)的化合物及其药学上可接受的盐:
[0010][0011]
r1、r2独立地表示选自如下的基团:
[0012]
氢;或nhc(o)nh-c1-6-烷基;或nhso
2-c1-6-烷基;或任选地被一个或多个r1a取代的nhc(o)nh-杂芳基;或杂芳基,其任选地被一个或多个选自氨基、c1-6-烷基、c(o)o-c1-6-烷基、腈、任选地被一个或多个r1a取代的杂芳基、nh-c1-6-烷基、nh-c1-6-杂环烷基、nh-c3-9-环烷基、任选地被一个或多个r1a取代的nh-杂芳基的基团取代;或nh-杂芳基,其任选地被一个或多个选自c1-6-烷基、c1-6-羟基烷基、c3-9-羟基杂环烷基、任选地被一个或多个r1a取代的杂芳基的基团取代;或nhc(o)-c1-6-烷基;或任选地被一个或多个r1a取代的nhc(o)-杂芳基;
[0013]
r1a表示选自如下的基团:
[0014]
卤素;腈;c1-6-烷基;c1-6-卤代烷基;c1-6-烷氧基;c1-6-卤代烷氧基;c(o)o-c1-6-烷基;c(o)oh;
[0015]
r3表示选自如下的基团:
[0016]
c1-6-烷基,其任选地被一个或多个选自r3a的基团取代;
[0017]
c1-3-烷二基-c3-6-环烷基,其任选地被一个或多个r3a取代;
[0018]
c1-3-烷二基-c3-6-杂环烷基,其任选地被一个或多个r3a取代;
[0019]
c3-6-杂环烷基,其任选地被一个或多个r3a取代;
[0020]
c3-6-环烷基,其任选地被一个或多个r3a取代;
[0021]
r3a表示选自氢;卤素;c1-2-烷基;羟基;c1-2-烷氧基的基团;
[0022]
r4表示选自如下的基团:
[0023]
c3-6-环烷基,其任选地被一个或多个r4a基团取代;或c1-6-烷二基-c3-6-环烷基,其任选地被一个或多个r4a基团取代;或c1-6-烷二基-c3-6-杂环烷基,其任选地被一个或多个r4a基团取代;
[0024]
r4a表示选自羟基;卤素;c1-2-烷基的基团。
[0025]
根据本发明的术语“药学上可接受的盐”包括式(i)化合物与药学上可接受的酸或碱的盐,特别是酸加成盐。以游离形式作为碱存在的式(i)化合物的酸加成盐形式可以通过用适当的酸处理该游离碱来获得,所述酸例如无机酸,例如氢卤酸如盐酸或氢溴酸、硫酸、硝酸、磷酸等;或有机酸,例如乙酸、三氟乙酸、草酸、羟基乙酸、丙酸、乳酸、丙酮酸、丙二酸、琥珀酸、马来酸、富马酸、苹果酸、酒石酸、柠檬酸、甲磺酸、乙磺酸、苯磺酸、对甲苯磺酸、环拉酸、水杨酸、对氨基水杨酸、双羟萘酸等。
[0026]
本发明还涉及式(i)化合物的所有立体异构体形式,例如对映异构体形式和非对映异构体形式或其混合物(包括所有可能的立体异构体混合物,例如外消旋体)。关于本发明,提及一种或多种化合物拟涵盖其每种可能的异构体形式及其混合物的化合物,具体提及特定的异构体形式除外。
[0027]
一些式(i)化合物也可以以互变异构体形式存在。这类形式尽管在上式中未明确显示但拟包括在本发明的范围内。
[0028]
应当理解,式(i)或本技术中所述的式中存在的每个单独原子实际上可以以其任何天然存在的同位素的形式存在,优选最丰富的同位素。因此,作为实例,式(i)或本技术中所述的式中存在的每个单独氢原子可以作为1h、2h(氘)或3h(氚)原子存在,优选1h。类似地,作为实例,式(i)或本技术中所述的式中存在的每个单独碳原子可以作为12c、13c或14c原子存在,优选12c。
[0029]
本发明在其范围内包括上述式(i)化合物的溶剂化物。这样的溶剂化物可以与常见的有机溶剂或水形成。
[0030]
本发明在其范围内还包括上述式(i)化合物的共晶。技术术语“共晶”用于描述中性分子组分以确定的化学计量比存在于结晶化合物内的情况。药物共晶的制备使得能够对活性药物成分的结晶形式进行改变,这又可以改变其物理化学性质而不损害其预期的生物活性(参见pharmaceutical salts and co-crystals,ed.j.wouters&l.quere,rsc publishing,2012)。
[0031]
根据本发明的化合物可以以不同的多晶型形式存在。尽管在上式中未明确显示,但这类形式拟包括在本发明的范围内。
[0032]
本发明在其范围内还包括上述式(i)化合物的前药。术语“前药”意指在体内代谢为本发明化合物或其盐的化合物。可以通过将前药施用于哺乳动物(例如大鼠、小鼠、猴或人)并鉴定例如血液或尿液中的化合物或其盐来鉴定该前药。
[0033]
在本发明的框架内:
[0034]
ct-z表示可以具有t至z个碳原子的碳链,例如可以具有1至7个碳原子的c1-7碳链;
[0035]
烷基为饱和的、直链或支链脂族基团;例如,c1-6-烷基表示直链或支链的1至6个碳原子的碳链,例如甲基、乙基、丙基、异丙基、丁基、异丁基、叔丁基、戊基、己基。烷基包括氘代基团,其中一个或多个氢原子被氘原子2h替代。
[0036]
烷二基为式c
nh2n
的二价直链或支链饱和烃基,例如-ch
2-ch
2-;
[0037]
烷基氨基是指在氨基上被取代的一个或多个烷基。作为烷基氨基的实例可以提及甲基氨基;乙基氨基;叔丁基氨基;二甲基氨基;
[0038]
羟基为-oh基团;
[0039]
羟基烷基为其中一个或多个氢原子被羟基取代的烷基;
[0040]
卤代烷基为其中一个或多个氢原子被卤原子取代的烷基;
[0041]
烷氧基,即-o-烷基基团;
[0042]
卤代烷氧基为其中一个或多个氢原子被卤原子取代的烷氧基;
[0043]
卤原子,即氟、氯、溴或碘原子;
[0044]
环烷基指单环或双环脂族基团,其可以包含双键,不是芳族的,并且在该基团中包含3至14个原子,优选3至9个原子。作为环烷基的实例可以提及环丙基;环丁基;环丁烯基;环戊基;环己基;螺十一烷基;螺[2.2]戊烷基;
[0045]
杂环烷基是指单环或双环饱和基团,在该基团中包含3至14个原子,优选3至10个原子,其可以包含双键,不是芳族的,并且其中一个或多个碳原子被选自氮、氧、硫的原子替代。作为杂环烷基的实例可以提及氮杂环丙烷基;吡咯烷基;哌啶基;氧杂环丁烷;氧杂-螺-十一烷基;
[0046]
羟基杂环烷基为其中一个或多个氢原子被羟基取代的杂环烷基;
[0047]
杂芳基是指单环或双环基团,其包含5至14原子,优选5至10个原子,其中该基团中至少一个环是芳族的,并且其中该基团中至少一个原子选自氮、氧、硫。作为杂芳基的实例可以提及三唑基;呋喃基;吡咯基;苯并二氢吡喃基;异喹啉基。
[0048]
根据一个实施方案,本发明的化合物选自式(i)的化合物,其中:
2-(2'-氨基-1,1'-联苯基)]钯(ii)
[0103]
ipa异丙醇
[0104]
conc.浓缩的
[0105]
scxscx-2丙磺酸官能化的二氧化硅
[0106]
tea三乙胺
[0107]
acoh乙酸
[0108]
aibn2,2
′‑
偶氮双(2-甲基丙腈)
[0109]
bedfordcatalyst氯(η
2-p,c-三(2,4-二叔丁基苯基)亚磷酸)(三环己基膦)钯(ii)
[0110]
nbsn-溴琥珀酰亚胺
[0111]
min分钟
[0112]
lcms方法
[0113]
方法1:
[0114]
x-bridgec18waters2.1x20mm,2.5μm柱
[0115]
柱温40℃
[0116]
流动相a:10mm甲酸铵水溶液+0.1%甲酸
[0117]
流动相b:乙腈+5%水+0.1%甲酸
[0118]
梯度程序:流速1毫升/分钟
[0119][0120]
方法2:
[0121]
流动相a:0.1%甲酸水溶液
[0122]
流动相b:0.1%甲酸的乙腈溶液
[0123]
phenomenex,kinetex-xbc18,2.1mmx100mm,1.7μm柱
[0124]
流速:0.6ml/min
[0125]
柱温:40℃
[0126]
注射体积:1μl
[0127][0128]
uv 215nm,pda光谱200
–
400nm,步长:1nm
[0129]
msd正扫描150-850
[0130]
方法3:
[0131]
x-bridge c18 waters 2.1x20mm,2.5μm柱
[0132]
柱温40℃
[0133]
流动相a:10mm甲酸铵水溶液+0.1%甲酸
[0134]
流动相b:乙腈+5%水+0.1%甲酸
[0135]
梯度程序:流速1ml/min
[0136][0137]
方法4:
[0138]
固定相:x-bridge c18 waters 2.1x20mm,2.5μm柱
[0139]
流动相a:10mm甲酸铵水溶液+0.1%氨溶液
[0140]
流动相b:乙腈+5%水+0.1%氨溶液
[0141]
流速:1ml/min
[0142][0143]
方法5:
[0144]
系统:waters classic acquity-qda,acquity pda
[0145]
固定相:waters acquity uplc beh,c18,2.1x50mm,1.7μm
[0146]
流动相a:10mm甲酸铵水溶液+0.1%氨溶液
[0147]
流动相b:乙腈+5%水+0.1%氨溶液
[0148]
流速:0.7ml/min
[0149]
温度:40℃
[0150][0151]
方法6:
[0152]
固定相:x-bridge c18 waters 2.1x20mm,2.5μm柱
[0153]
流动相a:10mm甲酸铵水溶液+0.1%甲酸
[0154]
流动相b:乙腈+5%水+0.1%甲酸
[0155]
流速:泵1:1ml/min,泵2:0.5ml/min
[0156][0157]
方法7:
[0158]
固定相:x-bridge c18 waters 2.1x20mm,2.5μm柱
[0159]
流动相a:10mm甲酸铵水溶液+0.1%氨溶液
[0160]
流动相b:乙腈+5%水+0.1%氨溶液
[0161]
流速:1ml/min
[0162][0163]
方法8:
[0164]
固定相:x-bridge c18 waters 2.1x20mm,2.5μm柱
[0165]
流动相a:10mm甲酸铵水溶液+0.1%氨溶液
[0166]
流动相b:乙腈+5%水+0.1%氨溶液
[0167]
流速:泵1:1ml/min,泵2:0.5ml/min
[0168][0169][0170]
方法9:
[0171]
固定相:x-bridge c18 waters 2.1x20mm,2.5μm柱
[0172]
流动相a:10mm甲酸铵水溶液+0.1%氨溶液
[0173]
流动相b:乙腈+5%水+0.1%氨溶液
[0174]
流速:1ml/min
[0175][0176]
中间体
[0177]
中间体1
[0178][0179]
2,3,6,7,8,9-六氢环戊二烯并[a]萘-1-酮
[0180]
在-78℃将1,2,3,4-四氢萘(5.17ml,37.8mmol)缓慢地加到alcl3(10.6g,79.4mmol)和丙-2-烯酰氯(3.4ml,41.6mmol)在dcm(300ml)中的混悬液中。随后让该混合物温热至室温过夜。谨慎地在冰上水解该溶液,分离有机相。用dcm将水相萃取2次,用碳酸钾水溶液洗涤合并的有机级分,用硫酸钠干燥。真空除去溶剂,通过柱色谱法纯化粗残余物,用0%至20%etoac的庚烷溶液梯度洗脱,得到标题化合物(1.01g,12%收率)。lcms[m+h]
+
187,rt 1.89min(方法1)。
[0181]
中间体2
[0182][0183]
2-羟基亚氨基-6,7,8,9-四氢-3h-环戊二烯并[a]萘-1-酮
[0184]
在0℃向中间体1(1.0g,5.36mmol)在乙醚(13ml)中的溶液中首先滴加饱和的乙醇hcl(0.22ml),然后滴加15%亚硝酸乙酯的乙醇溶液(4.81ml,7.62mmol)。在0℃ 30分钟后,通过过滤收集沉淀的产物,用乙醚洗涤,干燥,得到粗品标题化合物(1.15g,74%收率),其未经进一步纯化被用于下一阶段。lcms[m+h]
+
216,rt 1.77min(方法1)。
[0185]
中间体3
[0186][0187]
1,3-二氯-7,8,9,10-四氢苯并[h]异喹啉
[0188]
在0℃向中间体2(0.86g,3.99mmol)在pocl3(24ml)中的混悬液中加入pcl5(940mg,4.51mmol)。然后导入气态hcl,直至溶液饱和,将该反应体系在60℃搅拌4小时。然后再加入pcl5(316mg),在80℃持续搅拌2小时。真空除去溶剂,通过添加水缓慢地水解残余物,得到沉淀物,通过过滤采集该沉淀物。用水洗涤采集的固体,干燥,得到粗品标题化合物(1g,84%收率),其未经进一步纯化被用于下一阶段。lcms[m+h]
+
252,rt 2.50min(方法1)。
[0189]
中间体4
[0190][0191]
3-氯-7,8,9,10-四氢苯并[h]异喹啉
[0192]
将中间体3(0.88g,3.52mmol)、红磷(261mg,8.45mmol)在acoh(4ml)中的溶液和hi(57%)(1.59ml,12.1mmol)的混合物回流8小时。将热反应混合物过滤,蒸发,将残余物溶于水,通过添加浓nh4oh水溶液碱化。通过过滤采集沉淀物,将其溶于dcm,用盐水洗涤,用mgso4干燥,蒸发。通过柱色谱法纯化得到的粗残余物,用8%至40%etoac的庚烷溶液梯度洗脱,得到标题化合物(600mg,74%收率)。lcms[m+h]
+
218,rt 2.03min(方法1)。
[0193]
中间体5
[0194][0195]
3-氯-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰氯
[0196]
在密封试管中将中间体4(15.0g,68.9mmol)和氯磺酸(55ml,819mmol)的混合物在80℃加热3小时15min。然后用dcm(400ml)稀释该反应混合物,在0℃倾入h2o(400ml)。分离各相,用dcm(4x100ml)萃取水相,合并有机相,干燥(na2so4),过滤,真空浓缩,得到粗品标题化合物(21.8g,推定定量),其未经进一步纯化被用于下一步。lcms[m+h]
+
353(用ibunh2淬灭),rt 1.33min(方法4)。
[0197]
中间体6
[0198][0199]
3-氯-n-(2-氟-2-甲基-丙基)-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0200]
向在0℃搅拌的中间体5(21.8g,68.9mmol)在dcm(250ml)中的溶液中加入2-氟-2-甲基丙-1-胺hcl(9.85g,77.2mmol),然后加入dipea(30ml,172mmol)。然后将该反应体系在室温搅拌15小时。用h2o(200ml)、盐水(200ml)稀释该混合物,分离各相。用dcm(2x50ml)萃取水相,用h2o(150ml),然后用盐水(150ml)萃取合并的有机相,干燥,真空浓缩。将得到的固体与8:2的etoac:异己烷的混合物(100ml)一起研磨,得到标题化合物(19.7g,73%收率)。真空浓缩滤液,通过柱色谱法纯化,用0-50%etoac的异己烷溶液洗脱,得到第二批标题化合物(3.7g,12%收率)。lcms[m+h]
+
371,rt 1.23min(方法4)。
[0201]
中间体7&8
[0202][0203]
n-[3-氯-5-[(2-氟-2-甲基-丙基)氨磺酰基]-7,8,9,10-四氢苯并[h]异喹啉-10-基]氨基甲酸叔丁酯(7)
[0204]
n-[3-氯-5-[(2-氟-2-甲基-丙基)氨磺酰基]-7,8,9,10-四氢苯并[h]异喹啉-7-基]氨基甲酸叔丁酯(8)
[0205]
在50℃向中间体6(9.10g,24.5mmol)在etoac(235ml)中的溶液中加入nbs(4.85g,27.2mmol)和2,2'-偶氮双(2-甲基丙腈)(400mg,2.44mmol)。将该反应混合物在n2气氛中加热至90℃持续2.5小时。真空浓缩该混合物,将残余物溶于thf(200ml),用nh3气体吹扫10min,然后在70℃在密封容器中加热11小时。然后真空浓缩该混合物,在冰浴中混悬于dcm(200ml),然后向其中加入二碳酸二叔丁酯(7.9g,35mmol)在dcm(30ml)中的溶液,随后加入tea(9ml,64.6mmol)。4h后,再加入二碳酸二叔丁酯(1.0g,4.4mmol),将该反应体系搅拌3天。然后用盐水(200ml)、饱和nahco3(200ml)稀释该反应混合物,分离各相。用dcm(3x50ml)萃取水相,干燥合并的有机相,真空浓缩。通过柱色谱法纯化,用0-20%etoac的异己烷溶液
洗脱,得到标题化合物:
[0206]
中间体7(2.35g,19%收率);lcms[m+h]
+
486,rt 1.28(方法4)。
[0207]
中间体8(3.53g,27%收率);lcms[m+h]
+
486,rt 1.23(方法4)。
[0208]
中间体9
[0209][0210]
n-[3-环丙基-5-[(2-氟-2-甲基-丙基)氨磺酰基]-7,8,9,10-四氢苯并[h]异喹啉-7-基]氨基甲酸叔丁酯
[0211]
向中间体8(3.52g,6.52mmol)在甲苯(70ml)、1,4-二噁烷(40ml)和h2o(3.5ml)的混合物中的混悬液中加入环丙基硼酸(1.87g,21.8mmol)和磷酸三钾(3.95g,18.2mmol)。用n2吹扫该混合物5min,然后添加乙酸钯(ii)(90mg,0.40mmol)和四氟硼酸三环己基磷鎓(400mg,1.05mmol)。用n2吹扫得到的橙色混合物5min,然后在120℃加热11小时,此后再加入乙酸钯(ii)(90mg,0.40mmol)和四氟硼酸三环己基磷鎓(400mg,1.05mmol),再用n2吹该混合物5min,然后在120℃加热3天。真空浓缩该反应混合物,将残余物溶于etoac(400ml,包含几毫升ipa)。加入水(150ml)和盐水(200ml),分离各相。用etoac(100ml)萃取水相,干燥合并的有机相,真空浓缩,与et2o(150ml)一起研磨,得到标题化合物(2.20g,69%收率)。lcms[m+h]
+
492,rt 1.25min(方法4)。
[0212]
中间体10
[0213][0214]
7-氨基-3-环丙基-n-(2-氟-2-甲基-丙基)-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0215]
将盐酸(4n的二噁烷溶液,6ml)加到中间体9(500mg,0.98mmol),随后加入meoh(2ml),将得到的溶液在室温搅拌1.5小时。真空浓缩该反应混合物,通过10g scx柱,用7n nh3的meoh溶液洗脱,然后与et2o(20ml)一起研磨,得到标题化合物(356mg,93%收率)。lcms[m+h]
+
392,rt 1.32min(方法9)。
[0216]
中间体11
[0217][0218]
n-[3-环丙基-5-[(2-氟-2-甲基-丙基)氨磺酰基]-7,8,9,10-四氢苯并[h]异喹啉-10-基]氨基甲酸叔丁酯
[0219]
向中间体7(2.35g,4.84mmol)在甲苯(50ml)、1,4-二噁烷(20ml)和h2o(3ml)的混合物中的混悬液中加入环丙基硼酸(1.2g,14mmol)和磷酸三钾(2.6g,12mmol)。用n2吹扫该混合物5min,然后添加乙酸钯(ii)(55mg,0.24mmol)和四氟硼酸三环己基磷鎓(270mg,0.71mmol)。用n2吹扫得到的橙色混合物5min,然后在120℃加热9小时,此后再加入乙酸钯(ii)(55mg,0.24mmol)和四氟硼酸三环己基磷鎓(400mg,1.05mmol),再用n2吹该混合物5min,然后在120℃加热8小时。真空浓缩该反应混合物,溶于etoac(400ml),用盐水(150ml)洗涤。分离各相,用etoac(2x100ml)萃取水相。干燥合并的有机相,真空浓缩,与et2o(100ml)一起研磨,得到标题化合物(1.96g,82%收率)。lcms[m+h]
+
492,rt 1.29min(方法4)。
[0220]
中间体12
[0221][0222]
10-氨基-3-环丙基-n-(2-氟-2-甲基-丙基)-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0223]
将盐酸(4n的二噁烷溶液,6ml)加到中间体11(500mg,1.00mmol),随后加入meoh(2ml),将该溶液在室温搅拌1小时。真空浓缩该反应混合物,将其通过scx柱,用7n nh3的meoh溶液洗脱,然后与et2o(10ml)一起研磨,得到标题化合物(307mg,75%收率)。lcms[m+h]
+
392,rt 0.97min(方法4)。
[0224]
中间体13
[0225][0226]
3-氯-n-异丁基-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0227]
向中间体5(475mg,1.50mmol)在dcm(20ml)中的溶液中滴加2-甲基丙-1-胺
(0.74ml,7.51mmol),将该反应混合物在室温搅拌2小时。用dcm(50ml)稀释该反应混合物,用水洗涤。用mgso4干燥有有机层,减压浓缩。然后通过柱色谱法纯化残余物,用12%至60%etoac的庚烷溶液梯度洗脱,得到标题化合物(497mg,92%收率)。lcms[m+h]
+
353,rt 3.97min(方法2)。
[0228]
中间体14&15
[0229][0230]
7-溴-3-氯-n-异丁基-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺(14)
[0231]
10-溴-3-氯-n-异丁基-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺(15)
[0232]
将中间体13(150mg,0.42mmol)溶于etoac(15ml),加入nbs(75.6mg,0.42mmol),然后加入aibn(6.98mg,0.043mmol)。将不均匀混合物加热至90℃持续3小时。用etoac稀释反应混合物,用na2s2o3水溶液、水和盐水洗涤。用mgso4干燥有机层,减压浓缩。通过柱色谱法纯化粗残余物,用0%至50%etoac的庚烷溶液梯度洗脱,得到标题化合物(145mg,56%收率),为区域异构体混合物,纯度为71%。lcms[m+h]
+
431/433,rt 3.33min(方法3)[注意:共洗脱两种区域异构体相同rt]。
[0233]
中间体16&17
[0234][0235]
7-氨基-3-氯-n-异丁基-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺(16)
[0236]
10-氨基-3-氯-n-异丁基-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺(17)
[0237]
将中间体14&15的混合物(71%,145mg,0.24mmol)溶于7n氨的meoh溶液(2ml),在室温搅拌16小时。真空除去溶剂,加入dcm,随后加入水。分离有机层,用na2so4干燥,减压浓缩。通过柱色谱法纯化粗残余物,首先用5%至100%etoac的庚烷溶液梯度洗脱,以除去未溴化的原料,然后用0%至10%meoh的dcm溶液梯度洗脱,得到标题化合物(84mg,94%收率),为区域异构体混合物。lcms[m+h]
+
368,rt 2.09和2.14min(方法3)。
[0238]
中间体18&19
[0239][0240]
1-[3-氯-5-(异丁基氨磺酰基)-7,8,9,10-四氢苯并[h]异喹啉-7-基]-3-乙基-脲(18)
[0241]
1-[3-氯-5-(异丁基氨磺酰基)-7,8,9,10-四氢苯并[h]异喹啉-10-基]-3-乙基-脲(19)
[0242]
将异氰酸根合乙烷(19.8μl,0.25mmol)加到中间体16&17(84mg,0.22mmol)在dcm(4ml)中的混合物中。将该溶液在室温搅拌5小时,此后出现沉淀。加入meoh(5ml)。然后除去溶剂,通过酸性反相hplc纯化粗产物,得到标题化合物:
[0243]
中间体18(37mg,70%收率)。lcms[m+h]
+
439,rt 2.63min(方法3)。
[0244]
中间体19(24mg,44%收率)。lcms[m+h]
+
439,rt 2.70min(方法3)。
[0245]
中间体20&21
[0246][0247]
3-氯-n-异丁基-7-(甲磺酰氨基)-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺(20)
[0248]
3-氯-n-异丁基-10-(甲磺酰氨基)-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺(21)
[0249]
向中间体16&17(120mg,0.29mmol)在dcm(4ml)中的混合物中加入dipea(103.4μl,0.58mmol),随后加入甲磺酰氯(23μl,0.29mmol)。将该反应混合物在室温搅拌4小时。然后用dcm和水稀释该反应混合物。分离有机层,用na2so4干燥,减压浓缩。通过反相hplc(酸性条件)纯化,得到标题化合物:
[0250]
中间体20(65mg,98%收率)。lcms[m+h]
+
446,rt 2.68min(方法3)。
[0251]
中间体21(31mg,43%收率)。lcms[m+h]
+
446,rt 2.74min(方法3)。
[0252]
中间体22
[0253][0254]
1-[3-环丙基-5-[(2-氟-2-甲基-丙基)氨磺酰基]-7,8,9,10-四氢苯并[h]异喹
1.66(m,1h),1.19-1.00(m,7h),0.91-0.80(m,6h)。lcms[m+h]
+
445,rt 2.70min(方法2)。
[0265]
实施例3
[0266][0267]
1-[3-环丙基-5-(2-甲基丙基氨磺酰基)-7,8,9,10-四氢苯并[h]异喹啉-10-基]-3-乙基脲
[0268]
将中间体17(20mg,0.046mmol)和环丙基硼酸(7.8mg,0.091mmol)的混合物溶于氮气喷射过的二噁烷(2ml)。然后加入2m k2co3水溶液(68.3μl,0.23mmol),随后加入bedford催化剂(4.8mg,0.005mmol)。将该反应混合物在120℃在微波中在氮气气氛中搅拌2小时。减压浓缩该反应混合物,以除去二噁烷。用dcm(15ml)稀释残留的溶液,用水洗涤有机层,用mgso4干燥,减压浓缩。通过反相hplc(酸性条件)纯化,得到标题化合物(6mg,30%收率)。δh(500mhz,甲醇-d4)9.35(s,1h),8.31(s,1h),8.08(s,1h),5.82-5.49(m,1h),3.24-3.12(m,2h),3.06-2.98(m,1h),2.98-2.88(m,1h),2.63(d,j=6.9hz,2h),2.32-2.22(m,1h),2.21-2.10(m,1h),2.07-1.80(m,3h),1.71-1.55(m,1h),1.22-0.99(m,7h),0.82-0.76(m,6h)。lcms[m+h]
+
445,rt 2.78min(方法2)。
[0269]
实施例4
[0270][0271]
3-环丙基-7-(甲磺酰氨基)-n-(2-甲基丙基)-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0272]
将中间体16(63mg,0.14mmol)和环丙基硼酸(24.2mg,0.28mmol)的混合物溶于氮气喷射过的二噁烷(3ml)。然后加入2m k2co3水溶液(0.21ml,0.42mmol),随后加入bedford催化剂(15.1mg,0.014mmol)。减压浓缩该反应混合物,以除去二噁烷。用dcm(20ml)稀释残留的溶液,用水洗涤有机层,用mgso4干燥,减压浓缩。通过反相hplc(酸性条件)纯化,得到标题化合物(27mg,41%收率)。δh(500mhz,氯仿-d)9.35(s,1h),8.34(s,1h),8.22(s,1h),4.91-4.72(m,3h),3.32-3.22(m,1h),3.21-3.08(m,4h),2.94-2.70(m,2h),2.33-2.15(m,2h),2.11-1.95(m,3h),1.83-1.69(m,1h),1.19-1.13(m,2h),1.12-1.06(m,2h),0.91-0.84(m,6h)。lcms[m+h]
+
452,rt 2.89min(方法2)。
[0273]
实施例5
[0274][0275]
3-环丙基-10-(甲磺酰氨基)-n-(2-甲基丙基)-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0276]
将中间体17(30mg,0.067mmol)和环丙基硼酸(11.56mg,0.13mmol)的混合物溶于氮气喷射过的二噁烷(2ml)。然后加入2m k2co3水溶液(0.10ml,0.20mmol),随后加入bedford催化剂(7.1mg,0.007mmol)。将该反应混合物在120℃在微波中在氮气气氛中搅拌2小时。减压浓缩该反应混合物,以除去二噁烷。用dcm(15ml)稀释残留的溶液,用水洗涤有机层,用mgso4干燥,减压浓缩。通过反相hplc(酸性条件)纯化,得到标题化合物(10mg,33%收率)。δh(500mhz,氯仿-d)9.60(s,1h),8.18(s,1h),8.08(s,1h),5.59-5.53(m,1h),4.66(t,j=6.2hz,1h),4.54(d,j=7.3hz,1h),3.13(s,3h),3.07-3.00(m,1h),2.99-2.89(m,1h),2.71(t,j=6.6hz,2h),2.50-2.44(m,1h),2.24-2.18(m,1h),2.09-2.00(m,2h),1.98-1.89(m,1h),1.76-1.67(m,1h),1.20-1.13(m,2h),1.11-1.04(m,2h),0.85(d,j=6.7hz,6h)。lcms[m+h]
+
452,rt 2.94min(方法2)。
[0277]
实施例6
[0278][0279]
3-环丙基-n-(2-氟-2-甲基丙基)-7-[[6-(2-甲基四唑-5-基)吡啶-3-基]氨基]-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0280]
在室温向搅拌的中间体10(30mg,0.08mmol)在无水1,4-二噁烷(2.5ml)中的混悬液中加入5-溴-2-(2-甲基四唑-5-基)吡啶(37mg,0.15mmol)、叔丁醇钠(22mg,0.23mmol)和tbuxphos pd g3(9mg,0.01mmol)。用n2吹扫得到的混合物10min。再经过15min后,通过硅藻土过滤该反应混合物,真空浓缩。通过柱色谱法纯化,用0-20%meoh的etoac溶液洗脱,随后进行碱性反相柱色谱法,得到标题化合物(20mg,44%收率)。δh(300mhz,d6-dmso)9.48(s,1h),8.36(s,2h),8.22(d,j=2.7hz,1h),8.15(s,1h),7.88(d,j=8.6hz,1h),7.24(dd,j=8.8,2.8hz,1h),6.93(d,j=8.7hz,1h),4.95(d,j=8.6hz,1h),4.39(s,3h),3.39(s,1h),3.20(d,j=18.0hz,1h),2.96
–
2.79(m,2h),2.34
–
2.25(m,1h),1.97(s,4h),1.19(dd,j=21.4,3.2hz,6h),1.06(d,j=6.3hz,4h)。lcms[m-h]-549,rt 2.16min(方法8)。
[0281]
实施例7
[0282][0283]
3-环丙基-7-[3-[(2,5-二甲基吡唑-3-基)氨基]-1,2,4-三唑-4-基]-n-(2-氟-2-甲基丙基)-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0284]
向中间体22(55mg,0.10mmol)在dmf(1ml)中的溶液中加入甲酸酰肼(18mg,0.30mmol)和氯化汞(52mg,0.19mmol)。然后加入tea(40μl,0.29mmol),将该反应混合物在80℃加热4小时。然后通过硅藻土过滤该反应混合物(20ml mecn洗涤),真空浓缩。通过柱色谱法纯化,得到标题化合物(10mg,19%收率)。δh(300mhz,d6-dmso)9.51(s,1h),8.54(s,1h),8.35(s,1h),7.89(s,1h),7.75(s,1h),5.85(s,1h),5.67(s,1h),3.38(s,5h),2.78(d,j=20.0hz,2h),2.29(q,j=6.1hz,1h),2.20(s,2h),2.07(s,3h),1.98(s,2h),1.13(dd,j=21.4,4.9hz,6h),1.07(d,j=6.6hz,4h)。[一个h不可见]。lcms[m+h]
+
553,rt 2.14min(方法5)。
[0285]
实施例8
[0286][0287]
3-环丙基-n-(2-氟-2-甲基丙基)-10-[[6-(2-甲基四唑-5-基)吡啶-3-基]氨基]-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0288]
使用中间体12(30mg,0.08mmol)和相当化学计量的试剂,以与实施例6相同的方式合成。通过柱色谱法纯化,用0-20%meoh的etoac溶液洗脱,然后进行反相柱色谱法(碱性条件),得到标题化合物(6mg,14%收率)。δh(300mhz,d6-dmso)9.09(s,1h),8.43(s,1h),8.24(d,j=2.7hz,1h),7.99(s,1h),7.93(d,j=8.6hz,1h),7.36(dd,j=8.7,2.8hz,1h),7.02(d,j=8.4hz,1h),5.55(s,1h),4.40(s,3h),2.92(dd,j=19.6,9.9hz,4h),2.25
–
2.07(m,2h),1.94(s,1h),1.80(d,j=14.2hz,2h),1.24(d,j=21.4hz,6h),0.97(d,j=8.2hz,4h)[一个h不可见]。lcms[m-h]-549,rt 1.18min(方法6)。
[0289]
实施例9
[0290][0291]
5-氨基-1-[3-环丙基-5-[(2-氟-2-甲基丙基)氨磺酰基]-7,8,9,10-四氢苯并[h]异喹啉-7-基]咪唑-4-甲酸乙酯
[0292]
向2-氨基-2-氰基乙酸乙酯(15mg,0.07mmol,64%纯度)在mecn(1ml)中的溶液中加入原甲酸三乙酯(13μl,0.08mmol)。将得到的溶液在90℃加热1小时,然后冷却至室温。然后加入中间体10(20mg,0.05mmol),1小时后,真空浓缩该反应混合物,通过反相柱色谱法(酸性条件)纯化,得到标题化合物(9mg,31%收率)。δh(300mhz,d6-dmso)9.50(s,1h),8.40(s,1h),8.34(d,j=0.8hz,1h),7.65(s,1h),6.78(s,1h),6.24
–
6.09(m,2h),5.61(t,j=6.1hz,1h),4.18(q,j=7.1hz,2h),3.39(d,j=16.4hz,2h),2.81(dt,j=20.0,6.7hz,2h),2.35
–
2.28(m,1h),2.19
–
2.03(m,2h),1.92(d,j=10.2hz,2h),1.25(t,j=7.1hz,3h),1.16(d,j=21.4hz,6h),1.07(d,j=6.9hz,4h)。lcms[m+h]
+
530,rt1.68min(方法7)。
[0293]
实施例10
[0294][0295]
3-环丙基-n-(2-氟-2-甲基丙基)-7-[[5-(3-甲基-1,2,4-噁二唑-5-基)吡啶-3-基]氨基]-7,8,9,10-四氢苯并[h]异喹啉-5-磺酰胺
[0296]
使用中间体10(25mg,0.06mmol)和相当化学计量的试剂并且在100℃加热6小时,以与实施例6相同的方式合成。通过柱色谱法纯化,得到标题化合物(3mg,8%收率)。δh(300mhz,d6-dmso)9.49(s,1h),8.54(s,1h),8.47(d,j=1.8hz,1h),8.37(d,j=2.8hz,2h),8.16(s,1h),7.69(t,j=2.3hz,1h),6.92(d,j=8.8hz,1h),5.02(s,1h),3.43(d,j=18.9hz,2h),2.86(d,j=17.8hz,2h),2.43(s,3h),2.32(d,j=6.4hz,1h),1.97(s,4h),1.20(dd,j=21.4,5.5hz,6h),1.07(d,j=6.3hz,4h)。lcms[m+h]
+
551,rt 2.31min(方法8)。
[0297]
实施例11
[0298][0299]
1-[3-环丙基-5-[(2-氟-2-甲基丙基)氨磺酰基]-7,8,9,10-四氢苯并[h]异喹啉-7-基]-3-(2,5-二甲基吡唑-3-基)脲
[0300]
向搅拌的1,3-二甲基-1h-吡唑-5-胺(8mg,0.07mmol)在mecn(1ml)中的溶液中加入tea(20μl,0.14mmol)和1,1'-羰基二咪唑(11mg,0.07mmol)。1小时后,加入中间体10(25mg,0.07mmol)。再经过12小时后,用盐水(20ml)稀释该反应混合物,用dcm(3x20ml)萃取,干燥合并的有机层,真空浓缩。通过柱色谱法纯化,用0-30%meoh的etoac溶液洗脱,然后进行反相柱色谱法(酸性条件),得到标题化合物(3mg,9%收率)。δh(300mhz,d6-dmso)9.44(s,1h),8.38(s,1h),8.33(d,j=7.6hz,2h),8.18(s,1h),6.98(d,j=8.6hz,1h),5.93(s,1h),5.01(s,1h),3.54(s,3h),3.26(s,2h),2.92(d,j=18.9hz,2h),2.31(d,j=6.5hz,1h),2.07(s,3h),1.98(s,4h),1.21(dd,j=21.4,6.8hz,6h),1.06(d,j=6.8hz,4h)。lcms[m+h]
+
529,rt 1.88min(方法8)。
[0301]
实施例12
[0302][0303]
1-[3-环丙基-5-[(2-氟-2-甲基丙基)氨磺酰基]-7,8,9,10-四氢苯并[h]异喹啉-7-基]咪唑-4-甲酸乙酯
[0304]
向(z)-3-(二甲基氨基)-2-异氰基-丙-2-烯酸乙酯*(10mg,0.06mmol)在1-丁醇(1ml)中搅拌的溶液中加入中间体10(20mg,0.05mmol),将得到的混合物在150℃加热9小时。然后真空浓缩该混合物,通过柱色谱法纯化,用0-20%meoh的etoac溶液洗脱,然后进行反相柱色谱法,洗脱(碱性条件),得到标题化合物(4mg,15%收率)。δh(400mhz,d6-dmso)9.51(s,1h),8.38(s,1h),8.34(s,1h),7.85(d,j=1.3hz,1h),7.79(d,j=1.3hz,1h),7.60(s,1h),5.83(t,j=6.3hz,1h),4.17(qd,j=7.1,2.0hz,2h),3.45
–
3.39(m,1h),3.39
–
3.35(m,1h),2.88
–
2.69(m,2h),2.31
–
2.23(m,2h),2.22
–
2.11(m,1h),1.95(s,2h),1.22(t,j=7.0hz,3h),1.13(dd,j=21.3,2.4hz,6h),1.09
–
1.04(m,4h)。lcms[m+h]
+
515,rt 2.03min(方法8)。
[0305]
*根据wo2007/42545 a1,2007中的方法合成。
[0306]
体外生化测定:
[0307]
制备ige-tb试剂的方案
[0308]
将在100mm nahco3,ph 9.5中的172μm的86纳摩尔ige-fc(n265q,n371q)(young等人,1995)加到1mg lanthascreen
tm amine reactive tb chelate(thermofisher目录号pv3583)中,在20℃温育16小时。然后将材料缓冲液交换到磷酸缓冲盐水(为137mm nacl,2.7mm kcl,10mm na2hpo4,1.8mm k2hpo4,ph 7.4)中,将材料定量,通过测量280nm和343nm处的吸收测定tb缀合的程度。
[0309]
在s200 hr 10x300柱(ge healthcare)上通过分析型尺寸排阻色谱法测定了缀合材料的完整性。典型的缀合比为4:1 tb:ige-fc。
[0310]
young rj.,owens,rj.,mackay ga.,chan cmw.,shi j.,hide m.,francis dm.,henry aj.,sutton bj.和gould hj(1995)protein engineering 8:193-199
[0311]
制备sfcεr1α-y131a-af488试剂的方案
[0312]
使在100mm naoac ph 5.5中的400μm的400纳摩尔fcεr1α(y131a突变体)(cook等人,1997)与1mm终浓度的高碘酸钠(在100mm naoac中,ph 5.5)在22℃下反应60分钟。通过添加40μl乙二醇淬灭氧化并在22℃温育60分钟。将蛋白质缓冲液交换到缀合缓冲液(50mm nahco3,150mm nacl,ph 9.5)中并浓缩至750μm。
[0313]
将175纳摩尔蛋白质加入1mg alexa fluor
tm
488酰肼(invitrogen),并在22℃下温育16小时。加入氰基硼氢化钠(在缀合缓冲液中100mm)至终浓度为1mm,并在冰上温育60分钟。将蛋白质缓冲液交换到磷酸缓冲盐水(为137mm nacl,2.7mm kcl,10mm na2hpo4,1.8mm k2hpo4,ph 7.4)中,并将材料定量,并通过测量280nm和495nm处的吸收测定alexa fluor
tm
488缀合的程度。
[0314]
在s200 hr 10x300柱(ge healthcare)上通过分析型尺寸排阻色谱法测定了缀合材料的完整性。典型的缀合比为2:1 alexa fluor
tm
488:sfcεr1α
[0315]
cook jpd.,henry aj.,mcdonnell jm.,owens rj.,sutton bj.和gould hj(1997)biochemistry 36:15579-15588
[0316]
目的在于使用体外荧光共振能量转移(fret)测定法来测量ige-tb与受体的结合,以及化合物对结合的抑制。
[0317]
试剂
[0318]
使用的fret试剂为用铽标记的ige(fret供体)和用alexa fluor
tm 488标记的具有y131a突变的可溶性ige受体fcεrlα(fret接受体)。同时将未标记的fcεriα用于产生背景对照。测定缓冲液由20mm tris ph7.2、150mm nacl和0.002%吐温、1%dmso组成。
[0319]
测定反应
[0320]
根据以下程序进行了该测定法:在384孔半体积板中以25μl的体积进行每个测定反应。在dmso中以最终测定浓度(fac)的x50的浓度产生10点化合物连续稀释(3倍)。然后通过在测定缓冲液中稀释10倍的ige-tb制备化合物溶液。为了进行测定,将5μl稀释的化合物加入10μl,然后加入10μl fcεriα-y131a-af488。fret试剂facs为5nm ige-tb、25nm fcεr1α-y 131a-af488。通常,该测定法中化合物的最高fac为10μm。最终dmso浓度为2%。通过向fret试剂中加入5μl 1μm未标记的fcεriα(fac=200nm)测量了最小信号(min)。在包含fret试剂但不含化合物的孔中测量了最大fret信号(max)。
[0321]
在室温将本测定体系温育2小时,避光和防止蒸发,并适度搅拌。
[0322]
fret测量
[0323]
使用envision读板器(perkin elmer),通过在330nm处激发并在495/520nm处测量发射,进行了每孔fret的测量。
[0324]
在520处的发射/在495处的发射x 1000。
[0325]
fret比用于数据分析。
[0326]
数据分析
[0327]
z’计算如下(σ=标准偏差,且μ=平均值):
[0328]
1-((3xσ
max
)+(3xσ
min
))/(μ
max
–
μ
min
)
[0329]
高于0.5的z’被视为良好测定。
[0330]
从所有孔中减去背景信号(min)。使用减去背景的值,每个测试孔中化合物的抑制百分比计算如下:
[0331]
100
–
测试孔fret比/max fret比x 100。
[0332]
将抑制百分比对化合物浓度作图。使用xlfit5软件包,使用四参数逻辑拟合模型,测定了每个化合物的ic50值。
[0333]
结果如下:
[0334]
本发明的化合物显示19.8nm至5098nm范围的ic50值。下表显示了每个实施例的ic50值的范围:
[0335]
实施例编号fret ic
50
范围610-50纳摩尔浓度2,1050-100纳摩尔浓度1,3,4,7,8,9,11,120.1-1微摩尔浓度51-5微摩尔浓度
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1