病原微生物毒力基因关联模型及其建立方法和应用与流程
本发明涉及基因检测
技术领域:
,特别是涉及一种病原微生物毒力基因关联模型及其建立方法和应用。
背景技术:
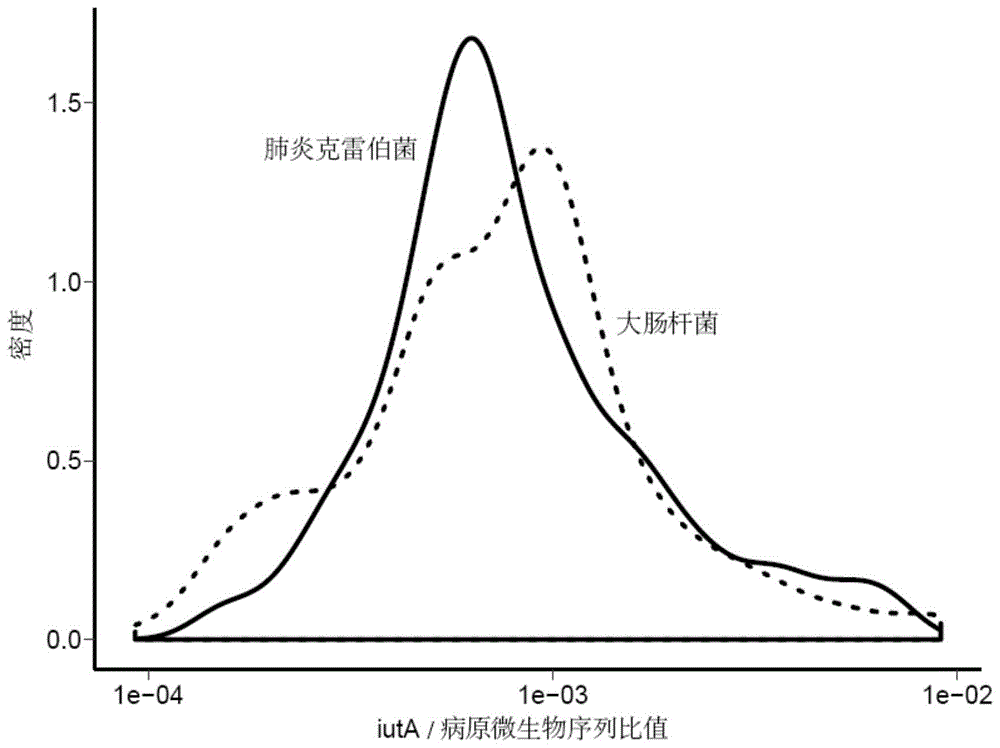
:毒力因子(virulencefactor,vfs)指由细菌,病毒,真菌等代谢产生的带有侵袭力和毒素等毒力性质的分子,表示病原体致病能力的强弱。主要用于微生物感染宿主时,在机体内定殖、突破机体的防御屏障、内化作用、繁殖和扩散,通过抑制或逃避宿主的免疫反应等出入宿主组织细胞,并从宿主获得营养及自身增殖生长的目的。细菌毒素按其来源、性质和作用等的不同,可分为外毒素和内毒素两大类。在大多数情况下,外毒素一般简称毒素。病原菌之所有能感染宿主并且在宿主环境中繁殖,通常就是依靠一系列的毒力因子之间相互协调作用起作用。所以研究病原菌的毒力因子,对于了解病原菌和宿主之间的相互作用,判断该病原菌的致病性强弱,为医生提供治疗指导意义非常重要。目前的毒力因子数据库是由中国医学科学院研发的病原菌毒力因子数据库vfdb(http://www.mgc.ac.cn/vfs/main.htm),研究还主要局限于肠道微生态科研方面,肠道微生态是由很多种微生物构成的群落,研究毒力因子一般用于判断细菌之间是否有群体失衡或者细菌功能紊乱。然而,目前并没有研究临床毒力因子方面的报道,并且vfdb数据库里面毒力基因众多,致病性强弱意义不同。因此,亟需开展对于临床常见病原微生物和毒力因子之间关联和影响的研究,获得临床常见致病病原微生物中对临床意义大的毒力基因(毒力基因是与生成毒力因子相关的基因),并且研究毒力基因与病原菌之间的比例关系,判断是什么病原菌,携带了什么毒力基因,及时准确地为医生提供判断该病原菌的毒性强弱,为临床治疗策略提供信息支撑。常规临床鉴定毒力因子,需要先培养出细菌,之后进行实验测试,比较复杂,周期较长且操作容易引入污染。也可基于固相芯片鉴定毒力因子,但和基于聚合酶链反应和微流控载体的毒力因子检测试剂盒的方法,只能检测特定的少量毒力因子,或者是少量细菌类型,并且无法把毒力因子与细菌对应起来,毒力因子可能来源于采样皮肤微生物污染或者由微生态细菌携带,导致结果受限且会带来假阳性。因此,采用传统方法检测毒力因子存在以下问题:检测方法需要时间长,方法操作繁琐、报告结果慢、通量低,往往仍需要先鉴定出菌种,才能进行毒力实验,而难以适应临床治疗需求;例如高毒力肺炎克雷伯菌常采用拉丝实验进行判断是否是高粘液表型(同时存在iuta和rmpa或者rmpa2毒力基因而形成的荚膜多糖高产的毒力因子),即需要肺克培养阳性才能进行实验判断。而基于固相芯片和微流控体系的毒力因子检测存在检测目标有限、成本高等特点,并且也不能确定细菌和毒力因子对应关系。病原宏基因组学(mngs)是一种不依赖于培养,直接从临床标本提取核酸并检测病原体的高通量测序技术。与传统临床的实验室检测方法相比,病原mngs是基于核酸水平对序列进行检测,可以突破不同病原体类型的局限性,无偏向性的全面覆盖数千种病原体,同时鉴定细菌、真菌、病毒和寄生虫等多种类型病原微生物,病原mngs已逐渐成为临床微生物鉴定领域的重要工具。技术实现要素:基于此,有必要针对上述问题,提供一种病原微生物毒力基因关联模型的建立方法,采用该方法,利用mngs的重要特点,是无偏差同时检测产毒力因子的毒力基因,可通过病原序列数和毒力基因序列数建立起病原-毒力因子关系,给临床提供准确判断病原体致病性强弱,及时为治疗和临床管理提供指导。一种病原微生物毒力基因关联模型的建立方法,包括以下步骤:建立病原微生物基因数据库:获取病原微生物的参考基因数据,构建病原微生物基因数据库;建立毒力基因数据库:获取毒力基因参考数据,构建毒力基因数据库;获取病原微生物和毒力基因数据:获取临床样本的病原微生物宏基因组测序数据,分别比对到上述病原微生物基因数据库和毒力基因数据库,得到每个临床样本中病原微生物的序列数据和该样本中毒力基因序列数据;建立毒力因子-病原微生物关联模型:对上述病原微生物的序列数据和毒力基因序列数据进行聚类分析,获得单个毒力基因与至少一个疑似关联病原微生物的正态分布模型,选取其中病原微生物丰度高,且毒力基因序列数与病原微生物序列数强相关的模型,即为病原微生物毒力基因关联模型。本发明人基于长期实践经验的基础上,经过调研总结和实验初筛后发现,毒力基因通常属于细菌基因组上的一段序列,因此,携带该毒力基因的菌株,毒力基因与细菌的比例是一个较为稳定的值,反映在病原宏基因组测序序列上,就是两者序列数的比值是较为稳定的一个范围。部分毒力基因由质粒携带,细菌群体里面携带毒力基因的质粒的占比,由于能量关系,也应该在一个比较稳定的比例,既不由于携带毒力基因过多造成能量的消耗,又能在应对当有外部刺激时,能够有足够比例的菌株能获得生存优势。基于上述理论和分析,本发明人基于本司已有5万例临床样品宏基因组测序数据展开分析,对细菌和毒力基因进行定量分析,使用高斯混合模型(正态分布)进行聚类获得细菌满足毒力关联的样品。由于不同病原体微生物体中包含的毒力基因的比例是不同的,每1000000条微生物序列对应于多少条毒力基因序列,受到不同细菌的基因组特征、细菌种类等因素的影响,反映在序列数比值分布上,就是不同均值、方差的正态分布。即本发明所述以最大似然概率和期望值最大算法对上述病原微生物的序列数据和毒力基因序列数据进行聚类分析,可获得单个毒力基因与至少一个疑似关联病原微生物的正态分布模型,再挑选丰度高,且毒力基因序列数与病原微生物序列数强相关的正态分布模型,即可判断样品中毒力基因是与何种微生物关联。可以理解的,上述病原微生物基因数据库和毒力基因数据库中各病原微生物种类和毒力基因,均可基于目前科学研究进展纳入,优选具有明确临床意义的且重要的病原微生物和毒力基因。对于病原微生物的参考基因数据,可从ncbi、vfdb等权威数据库中获取。可以理解的,本发明的方法可应用于细菌和/或真菌中。在其中一个实施例中,所述毒力基因及毒力基因的参考数据由vfdb数据库获取。可以理解的,上述数据也可根据研究进展,从其它可靠、权威数据库中获取。在其中一个实施例中,所述建立毒力因子-病原微生物关联模型步骤中,通过以下方法进行聚类分析:计算毒力基因序列与病原微生物序列的对数比值,对该对数比值用高斯混合模型进行聚类分析,具体过程为:先以期望值最大算法进行迭代,对于每个样品分别计算该毒力基因由该混合模型内的每种病原微生物携带的概率,获得每种病原微生物携带该毒力基因的先验概率,并获得每种病原微生物携带该毒力基因时,测序观察到毒力序列的条件概率;然后使用最大似然概率算法计算每个样品在该毒力基因序列和病原微生物序列观察值下,该毒力基因最可能是属于哪种病原微生物分类,从而实现聚类。在其中一个实施例中,所述毒力基因及毒力基因的参考数据由vfdb数据库获取。在其中一个实施例中,所述建立毒力因子-病原微生物关联模型步骤中,在获得病原微生物毒力基因关联模型后,还对该模型进行筛选质控,保留同时符合以下条件的模型:1)符合该模型的临床样本数≥30个;2)该模型中作为来源归属的病原微生物丰度中位数≥1000条序列;3)所述毒力基因序列数与病原微生物序列数强相关的条件为:pearson相关系数,spearman相关系数,线性系数均具有统计学意义的显著性,且相关系数cor≥0.6;4)该模型中毒力基因序列数与病原微生物序列数比值密度分布在log维度下为正态分布。上述对各种条件的筛选质控中,由于需要有足够的检出样品数才能保证结果的稳定性,因此限定符合该模型的临床样本数≥30个。由于细菌的平均基因组长度为5000,000bp左右,毒力基因长度平均在1000bp左右,因此有足够丰度的细菌序列检出才能保证毒力基因检出序列是可靠的,因此,可限定作为关联的病原微生物丰度中位数应≥1000条。在其中一个实施例中,所述获取病原微生物和毒力基因数据步骤中,获取临床样本的病原微生物宏基因组测序数据后,先去除低质量测序数据,再比对到人参考基因组,去除人源序列,得到非人基因组序列,按照以下方法分析数据:分析得到病原微生物的序列数据:将获得的非人基因组序列比对到所述病原微生物基因数据库;根据比对序列数进行病原微生物丰度的定量计算,并解读判断每个临床样本检出的病原体微生物,获得病原微生物的序列数据;分析得到毒力基因的序列数据:将获得的非人基因组序列比对到所述毒力基因数据库,根据比对序列数进行毒力基因丰度的定量计算,获得毒力基因的序列数据。在其中一个实施例中,所述获取病原微生物和毒力基因数据步骤中:对于每个毒力基因按基因长度1000±100bp进行标准化处理,如该毒力基因对应多个参考序列版本,则选择标准化后丰度最高的版本作为该毒力基因的丰度。在其中一个实施例中,所述建立毒力因子-病原微生物关联模型步骤中,还对所得病原微生物毒力基因关联模型按照正态分布95%置信区间的计算方法,获得对应的毒力基因与病原微生物比例的95%置信区间。可以理解的,上述模型建立方法适用的样本类型广泛,包括但不限于肺泡灌洗液、痰液、咽拭子,以及血液、脑脊液等,病原宏基因(宏转录)组的样本类型的适用中。本发明还公开了上述的病原微生物毒力基因关联模型的建立方法得到的病原微生物毒力基因关联模型。本领域技术人员知晓,毒力因子为临床病原菌产生的对人体有毒性或者较大毒性的因素,而毒力基因是生成毒力因子相关的基因,一种毒力因子可能由单个或者多个毒力基因共同作用而产生。在其中一个实施例中,病原微生物与毒力基因和毒力因子的关联关系如下所示:细菌名称毒力基因或其组合毒力因子名称金黄色葡萄球菌lukf-pv或luks-pvpvl化脓性链球菌speaspea肺炎克雷伯菌iuta和rmpa组合,或iuta和rmpa2组合hvkp艰难梭菌toxa或toxbtcda/tcdb肠聚集性大肠杆菌aggraggr肠致病性大肠杆菌bfpabfpa肠致病性大肠杆菌eaeeae肠产毒素性大肠杆菌elta或eltbelt肠产毒素性大肠杆菌estiaest志贺毒性大肠杆菌stx1a或stx1bstx1志贺毒性大肠杆菌stx2a或stx2bstx2肠侵袭性大肠杆菌ipahipah。本发明人在充分调研、研究临床病原微生物感染及治疗情况后,筛选了具有明确临床意义的且重要的毒力因子列表,包括pvl、spea、iuta、rmpa/rmpa2、tcda/tcdb、aggr、bfpa、eae、elta/eltb、est、stx1、stx2等。得到病原微生物毒力基因关联模型,明确上述毒力因子与病原微生物的关联关系,其中各毒力因子临床意义如下:pvl是金黄色葡萄球菌常见毒力因子之一,常引起白细胞坏死或坏死性肺炎;pvl其包含两个由pvl基因编码的亚基,luks-pv和lukf-pv,且两个亚基单独均可产生毒力。spea,一种超级抗原,是由化脓性链球菌分泌的促红细胞毒素,其通过非特异性激活t细胞并刺激炎性细胞因子的产生来诱导炎症,易导致猩红热和链球菌中毒性休克综合征。iuta、rmpa/rmpa2是由phvkp质粒编码的毒力基因,是高毒力肺炎克雷伯菌的分子标志物;与传统的肺炎克雷伯菌(ckp)感染的流行病学特征相比,高毒力肺炎克雷伯菌(hvkp)易在社区引起多部位的组织侵袭性感染。tcda/b是一种由艰难梭菌编码产生的毒素,tcda表现为为肠毒素,tcdb表现为细胞毒素;其易导致肠组织损伤,引起腹泻、假膜性结肠炎甚至会危及生命。aggr是大肠杆菌中聚集黏附菌毛i转录激活因子,是肠聚集性大肠杆菌分子标签之一;肠聚集性大肠杆菌易引起急性、持续性腹泻和水样分泌性腹泻。bfpa是iv型成束菌毛(bfp)主要成份,该菌毛参与宿主细胞黏附和毒力作用,是肠致病性大肠杆菌的分子标签之一;肠致病性大肠杆菌易引起婴幼儿急性腹泻或慢性腹泻。eae是大肠杆菌产生黏附的必要成份,是肠致病性大肠杆菌的分子标签之一;肠致病性大肠杆菌易引起婴幼儿急性腹泻或慢性腹泻。elt是一种由大肠杆菌表达的不耐热肠毒素,是肠产毒素性大肠杆菌的分支标签之一;肠产毒素性大肠杆菌易导致发烧、恶心、水样腹泻并伴有腹部绞痛等症状。est是一种由大肠杆菌表达的热稳定肠毒素,是肠产毒素性大肠杆菌的分支标签之一;肠产毒素性大肠杆菌易导致发烧、恶心、水样腹泻并伴有腹部绞痛等症状。stx1志贺毒素1型,和细胞膜特异性受体结合,摄入志贺毒素会产生腹痛和水样腹泻症状,严重危机生命,是志贺毒性大肠杆菌的分子标签之一。stx2志贺毒素2型,和细胞膜特异性受体结合,摄入志贺毒素会产生腹痛和水样腹泻症状,严重危机生命,是志贺毒性大肠杆菌的分子标签之一。ipah是大肠杆菌携带的侵袭性质粒编码的蛋白,其利用黏附素蛋白去结合并进入肠细胞,可引起肠壁损坏,导致大量腹泻和高热,是肠侵袭性大肠杆菌的分子标签之一。本发明还公开了上述的病原微生物毒力基因关联模型在病原微生物毒力基因检测中的应用。本发明还公开了一种病原微生物毒力基因检测装置,包括:存储装置,用于存储上述的病原微生物基因数据库和毒力基因数据库;分析装置,用于获取待测样本的病原微生物宏基因组测序数据,并按照上述方法分析得到该样本中病原微生物的序列数据和所对应的毒力基因序列数据;并对病原微生物毒力基因关联模型按照正态分布95%置信区间的计算方法,获得对应的毒力基因与病原微生物比例的95%置信区间,设为阈值判断区间;将待测样本中毒力基因序列和病原微生物序列的比值与所述阈值判断区间进行比较,如该待测样本中毒力基因序列和病原微生物序列的比值落入所述阈值判断区间,则判断该毒力基因与此模型的病原微生物具有关联;输出装置,用于输出上述判断结果。可以理解的,上述各对应的毒力基因与病原微生物比例的95%置信区间,可以预先根据大样本量的数据计算得到而预存,在进行个别临床样本的毒力基因检测时,直接调用比较即可。本发明还公开了一种病原微生物毒力基因检测方法,包括以下步骤:检测:取待测样本,进行病原微生物宏基因组检测,获得测序数据;分析:按照上述方法分析得到该样本中病原微生物的序列数据和所对应的毒力基因序列数据;并对病原微生物毒力基因关联模型按照正态分布95%置信区间的计算方法,获得对应的毒力基因与病原微生物比例的95%置信区间,设为阈值判断区间;将待测样本中毒力基因序列和病原微生物序列的比值与所述阈值判断区间进行比较,如该待测样本中毒力基因序列和病原微生物序列的比值落入所述阈值判断区间,则判断该毒力基因与此模型的病原微生物具有关联。与现有技术相比,本发明具有以下有益效果:本发明的一种病原微生物毒力基因关联模型的建立方法所得模型,可用于检测毒力基因,特别是可用于将毒力基因与相应的病原微生物建立关联关系,给临床提供准确判断病原体致病性强弱,及时为治疗和临床管理提供指导。并且具有无需培养,采样需求小,样本类型无限制,检出时间短,检出毒力基因全面的优势。附图说明图1为实施例1中iuta序列与病原微生物序列比值模型示意图;图2为标准正态分布的95%置信区间示意图;图3为实施例2中检出肺炎克雷伯菌的样品的归属模型示意图;图4为实施例2中iuta基因与肺炎克雷伯菌的比值分布的盒子图。具体实施方式为了便于理解本发明,下面将参照相关附图对本发明进行更全面的描述。附图中给出了本发明的较佳实施例。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的
技术领域:
的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。以下实施例所用试剂,如非特别说明,均为市售可得。实施例1一种病原微生物毒力基因关联模型的建立方法,包括以下步骤:1、建立病原微生物基因数据库。获取病原微生物的参考基因数据,构建病原微生物基因数据库,可按常规方法进行,本实施例中,采用本司申请号为201910779825.0的中国发明专利所公开技术方案构建得到的病原微生物基因组数据库进行后续分析比对。可以理解的,本发明的方法并不受限于病原微生物的具体数据库,开源通用的数据库均可。2、建立毒力基因数据库。2.1调研获得临床意义明确的毒力基因列表。基于目前科学研究进展,收集具有明确临床意义的且重要的毒力基因列表,包括pvl、spea、iuta、rmpa/rmpa2、tcda/tcdb、aggr、bfpa、eae、elta/eltb、est、stx1、stx2等。2.2建立毒力基因数据库本实施例中,从vfdb数据库http://www.mgc.ac.cn/vfs/download.htm下载毒力因子参考序列文件,并使用coredataset(核心集)用bwa软件建立比对的索引文件,建立毒力基因数据库。3、获取病原微生物和毒力基因数据。3.1数据处理。获取临床样本的病原微生物宏基因组测序(mngs)数据,首先使用fastp软件去除接头、低质量或者长度小于35bp的序列;然后用bwa软件比对到人参考基因组,去除比对到人参考基因上的序列;之后,获得非人基因组序列(unhostreads)分别比对到上述病原微生物基因数据库和毒力基因数据库,得到每个临床样本中病原微生物的序列数据和所对应的毒力基因序列数据。3.2病原微生物定量分析流程。将上述非人基因组序列用bwa比对到上述病原微生物基因数据库(包含18562种微生物的参考序列);根据比对序列数进行微生物丰度的定量以及解读判断每个临床样品的病原体微生物。3.3毒力基因定量分析流程。将上述非人基因组序列用bwa比对到毒力数据库;根据比对序列数进行毒力基因的丰度的定量以及覆盖度计算;最后,对于每个毒力基因按基因长度1000bp进行标准化处理,如果一种毒力基因有多个参考序列版本,考虑到每个临床样本里面,每种毒力基因通常只包含其中一种序列版本,表现在样品的比对reads数上,就是丰度最高的那个参考版本,因此选择标准化后丰度最高的版本作为该毒力基因的丰度及覆盖度计算参考,示例说明如下:用bwa软件将序列比对到毒力数据库,对于每条序列,如果比对到毒力基因的比对率大于90%且错配率小于8%,则认为该序列比对上了该毒力基因;然后统计每种毒力基因比对到的序列数,以及序列覆盖到毒力基因的区域比例(覆盖度);最后,由于每种毒力基因可能有多个版本的参考序列,基因长度不同,为了方便模型计算,统一标准化到1000bp,即以比对符合序列数除以该版本基因长度数值,再乘以1000,比如毒力基因a有两个版本a1和a2,其中a1的长度为600bp,比对到了5条序列,a2的长度为800bp,比对到了7条序列,那么按1000bp标准化来计算,a1比对到的序列值为5/600×1000=8.33,a2比对到的序列值为7/800×1000=8.75,因此,a基因最终的丰度为8.75(选择标准化后值最大的版本)。4、建立毒力因子-病原微生物关联模型。对上述病原微生物的序列数据和毒力基因序列数据进行聚类分析计算毒力基因序列与病原微生物序列的对数比值,对该对数比值用高斯混合模型进行聚类分析,具体过程为:先以期望值最大算法进行迭代,对于每个样品分别计算该毒力基因由该混合模型内的每种病原微生物携带的概率,获得每种病原微生物携带该毒力基因的先验概率,并获得每种病原微生物携带该毒力基因时,测序观察到毒力序列的条件概率;然后使用最大似然概率算法计算每个样品在该毒力基因序列和病原微生物序列观察值下,该毒力基因最可能是属于哪种病原微生物分类,从而实现聚类。获得单个毒力基因与至少一个疑似来源病原微生物的正态分布模型。以下以iuta毒力基因为例进行说明。本实施例中,首先计算毒力基因iuta和病原微生物的序列比例,并进行log转换,然后用r软件mclust包对这个log转换后的比例进行高斯正态分布模型分类。具体过程为:先以期望值最大算法进行迭代,对于每个样品分别计算该毒力基因由该混合模型内的每种病原微生物携带的概率,获得每种病原微生物携带该毒力基因的先验概率,并获得每种病原微生物携带该毒力基因时,测序观察到毒力序列的条件概率;然后使用最大似然概率算法计算每个样品在该毒力基因序列和病原微生物序列观察值下,该毒力基因最可能是属于哪种病原微生物分类,从而实现聚类。经过上述分析发现,iuta毒力基因可能来源于大肠杆菌、肺炎克雷伯菌、福氏志贺菌等3种不同情况,如图1所示,iuta分别来源于大肠杆菌、肺炎克雷伯菌、福氏志贺菌3种不同情况的正态分布。之后挑选其中病原微生物丰度高且毒力基因reads与细菌reads线性最相关(即强相关),由于可能存在强影响点对线性模型的影响,计算线性相关时使用的是r软件的robustbase稳健回归模型,保证结果的稳健性。通过上述分析,可认定iuta毒力基因来源于肺炎克雷伯菌,得到iuta-肺炎克雷伯菌归属模型。对上述获得的类(iuta-肺炎克雷伯菌关联模型)进行质控,符合以下要求1)符合该模型的临床样本数≥30个,符合质控要求;2)该模型中作为来源归属的病原微生物丰度中位数≥1000bp,符合质控要求;3)所述毒力基因序列数与病原微生物序列数强相关的条件为:pearson相关系数,spearman相关系数,线性系数均显著p≤0.05,且相关系数cor≥0.6,符合质控要求;4)该模型中毒力基因序列数与病原微生物序列数比值密度分布在log维度下为正态分布。该模型中样品里面肺炎克雷伯菌的序列reads中位数不能小于1000,毒力基因reads数与肺炎克雷伯菌reads数线性相关显著且pearson和spearman相关系数均大于0.6,为强相关,符合质控要求;上述iuta-肺炎克雷伯菌关联模型的高斯正态分布图如图1所示,从图上进行观察也可看出比例分布是正态分布形态。按照上述方法,对具有明确临床意义的且重要的毒力因子及其所对应的毒力基因进行分析,建立模型,得到病原微生物与毒力基因和毒力因子的关联关系如下表所示:表1.病原微生物与毒力因子的关联关系细菌名称毒力基因或其组合毒力因子名称金黄色葡萄球菌lukf-pv或luks-pvpvl化脓性链球菌speaspea肺炎克雷伯菌iuta,rmpa或iuta,rmpa2hvkp艰难梭菌toxa或toxbtcda/tcdb肠聚集性大肠杆菌aggraggr肠致病性大肠杆菌bfpabfpa肠致病性大肠杆菌eaeeae肠产毒素性大肠杆菌elta或eltbelt肠产毒素性大肠杆菌estiaest志贺毒性大肠杆菌stx1a或stx1bstx1志贺毒性大肠杆菌stx2a或stx2bstx2肠侵袭性大肠杆菌ipahipah。5、毒力基因与细菌比例置信区间。按照如图2所示标准正态分布的95%置信区间方式,计算上述iuta-肺炎克雷伯菌关联模型的正态分布95%置信区间,也即是log维度下的均值±1.96倍方差,即为iuta基因与肺炎克雷伯菌的比例的95%置信区间,为[5.4e-5,3.7e-3],对应于单个肺炎克雷伯菌可以携带0.29到19.56个iuta毒力基因序列实施例2从5万例临床样品里面,挑选760例检出肺炎克雷伯菌的样品,首先计算毒力基因iuta和肺炎克雷伯菌klebsiellapneumoniae的序列比例,并进行log转换。以实施例1的iuta-肺炎克雷伯菌关联模型把iuta与肺炎克雷伯菌klebsiellapneumoniae的序列比例分为3类(a,b,c),如图3所示,其中b类是满足模型的样品(来源于肺炎克雷伯菌),a类可能是来自大肠杆菌、福氏志贺菌的样品,c类是无iuta序列检出的样品;图4为iuta基因与肺炎克雷伯菌的比值分布的盒子图,其中上下两条虚线之间是满足模型的样品比值分布的95%置信区间,具体为[5.4e-5,3.7e-3]。我们后续应用时,即可以根据iuta与肺炎克雷伯菌序列的比值是否落在该区间来判断该iuta毒力基因是否来源于肺炎克雷伯菌(即携带iuta的肺炎克雷伯菌)。如果一个临床样品,同时判断是携带iuta和rmpa毒力基因的肺炎克雷伯菌,则判断该样品为产hvkp高毒力因子的肺炎克雷伯菌。实施例3按照上述实施例1的方法,根据5万例临床样品数据,建立多种病原微生物毒力基因关联模型,并得到如下各毒力基因与细菌比例95%置信区间,用于临床判断应用。表2.毒力基因与细菌比例95%置信区间毒力基因细菌95%下置信比例95%上置信比例bfpa肠致病性大肠杆菌0.00001820.001818652tcda艰难梭菌0.00002330.002330865tcdb艰难梭菌0.00002330.002330865eae肠致病性大肠杆菌0.00001820.001818652elta肠产毒素性大肠杆菌0.00001820.001818652eltb肠产毒素性大肠杆菌0.00001820.001818652estia肠产毒素性大肠杆菌0.00001820.001818652iuta肺炎克雷伯菌0.00005430.003666461lukf-pv金黄色葡萄球菌0.00008310.000612467luks-pv金黄色葡萄球菌0.000220730.000387329rmpa肺炎克雷伯菌0.0001086510.000532625rmpa2肺炎克雷伯菌0.00005970.002014863spea化脓性链球菌0.0000540.005398306stx1a志贺毒性大肠杆菌0.00001820.001818652stx1b志贺毒性大肠杆菌0.00001820.001818652stx2a志贺毒性大肠杆菌0.00001820.001818652stx2b志贺毒性大肠杆菌0.00001820.001818652实施例4临床分离株模型验证应用。1、样本提取获得100例临床样品,所得样本按照以下流程提取核酸:按照北京天根生化有限公司微量样本基因组dna提取试剂盒(dp316)方法,提取模拟临床样本的dna,核酸提取步骤包括:1)上述预处理后得到的600μl样品,加入到含玻璃珠破壁管中,采用物理震荡仪进行物理破壁。2)短暂离心后,取300μl样品到新的1.5ml管中,加10μlproteinasek溶液,加入100μl预混carrierrna(浓度1μg/μl)的gb,轻轻颠倒混匀,短暂离心以去除管盖内壁的液滴。3)56℃温浴10min,并不时轻摇样品。4)在样品中加入从-20℃冻存的无水乙醇。轻轻颠倒混匀样品,室温放置3min。短暂离心以去除管盖内壁的液滴。5)将上一步所得溶液都加到一个吸附柱cr2中,12000rpm离心30sec,弃废液,将吸附柱cr2放回收集管中。6)向吸附柱cr2中加入500μl缓冲液gd,12000rpm离心30sec,弃废液,将吸附柱cr2放回收集管中。7)向吸附柱cr2中加入600μl缓冲液pw,12000rpm离心30sec,弃废液,将吸附柱cr2放回收集管中。8)重复操作步骤7)。9)12000rpm离心2min,倒掉废液。将吸附柱cr2置于室温放置2-5min,以彻底晾干吸附材料中残余的漂洗液。10)将吸附柱cr2转入一个干净的离心管中,向吸附膜中间位置悬空滴加50μl洗脱缓冲液tb,室温放置2-5min,12000rpm离心2min,将溶液收集到离心管中。11)采用qubit3.0荧光定量仪(thermofisher)准确定量dna样本浓度。2、文库构建。1)预先取出﹣20℃保存的5×ttbl和ttemixv1,4℃融化,充分混匀后短暂离心。取出4℃保存的磁珠,室温平衡30min,充分振荡混匀后短暂离心备用。2)dna片段化:按照4μl5xttbl,1ngdna,5μlttemixv1,补齐ddh2o至20μl,配置反应体系;在pcr中运行如下反应程序:105℃热盖,55℃10min,10℃保存。3)反应完成后立即向产物中加入5μl5xts,使用移液器轻轻吹打充分混匀,置于室温放置5min。4)pcr富集:按照如下体系配置,25μl步骤3产物,10μl5xtab,上下游引物各5μl,1μltae,使用移液器轻轻吹打混匀,将反应管置于pcr仪中,运行如下反应程序:105℃热盖,72℃3min,98℃30sec,此后98℃15sec,60℃30sec,72℃3min进行5-15个循环,72℃5min,4℃保存。5)pcr反应后进行扩增产物长度分选。6)产物分选:将提前室温平衡好的磁珠,按照第一轮0.7x,第二轮磁珠用量0.15x分选平均长度为350bp的文库片段。提取后的核酸样本按照诺维赞建库试剂盒indexueprepdnalibraryprepkitv2forillumina(td503-01)说明书进行dna文库构建。3、上级测序。构建好的核酸文库采用illuminanextseq550进行测序。每个样本测序20mreads数据量。4、测序数据分析。按照实施例1的方法进行病原微生物和毒力基因进行定量分析。5、病原体与毒力因子判断通过首先解读判断细菌是否是真实病原体,对于检出毒力基因的样本,判断毒力基因与细菌的序列比例模型关系,判断毒力因子是否为真:毒力基因与病原体序列比值是否落在模型获得的95%置信区间范围内,且满足检出的毒力因子条件(比如hvkp毒力因子需要同时判断iuta和rmpa/rmpa2都满足模型),结果如下表所示:表3.100例临床样本毒力因子检出注:b开头编号样品为血液样品,m开头编号样品为肺泡灌洗液样品,最后一列是根据毒力基因结果的模型判断的毒力因子,未知指无法与毒力因子模型匹配(大部分是单独检出iuta或者rmpa的肺炎克雷伯菌株)。上述结果表明,100例临床样品判断出5例高毒力菌株,经与临床获知患者情况相对比,较为一致。以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1