一种基于字典学习的氨基酸序列特征提取方法
1.本发明属于数据挖掘技术领域,涉及一种基于字典学习的氨基酸序列特征提取方法。
背景技术:
2.随着人类基因组计划的顺利进展,越来越多的蛋白质被测定出来,而通过实验确定其结构与功能的蛋白质则相对较少,且费时、费力、费财,实验中可能还会遇到一些目前无法解决的困难,因此探索利用理论计算方法来研究蛋白质结构和功能具有重要意义。如何从一条氨基酸序列提取它的有用信息,并用适当的数学方法来描述或表示这些信息,使之能正确反映序列与结构或功能之间的关系,对于蛋白质分类研究是至关重要的,也是决定分类质量的关键。目前的氨基酸序列的特征提取方法主要分为两类:一类为仅仅基于氨基酸组成和位置的方法;另一类为基于氨基酸物理化学性质的方法。氨基酸是组成蛋白质的基本单位,一条蛋白质包含的基本信息是20种氨基酸的种类和排列顺序,因此基于氨基酸组成和位置的特征提取算法是最简单、最直观的方法,主要有氨基酸组成(aac)、熵密度(edp)、n阶耦联组成(n
‑
occ)和完全信息集(cis)。氨基酸的侧链决定了氨基酸的种类、20种氨基酸侧链在形状、大小、负电性、水性以及酸碱性等方面都存在差异,正是这20种氨基酸的差异,使各种不同组合的氨基酸序列形成各种不同的蛋白质结构,并适应各类环境,完成其特定的生理功能。蛋白质的生物学活性和理化性质主要决定其空间结构的完整,因此仅仅知道蛋白质的氨基酸组成和它们的排列顺序并不能完全了解蛋白质的结构,需要考虑氨基酸的性质。目前,基于氨基酸物理化学性质特征提取方法主要有自相关函数、伪氨基酸组成(pseaa)、准序列次序作用和疏水模式组成(hp)。
3.上述所介绍的方法会丢失氨基酸的顺序信息以及其间的相互作用或计算量非常大等局限性,对于进一步预测蛋白质结构类起到的效果并不是十分明显。
技术实现要素:
4.本发明的目的是针对现有预测蛋白质结构类的需求,提出一种基于字典学习的氨基酸序列特征提取方法。该方法使用aaindex作为初始特征来分析序列,aaindex是一个包含aaindex1、aaindex2、aaindex3的数字指标数据库,它代表氨基酸和对氨基酸的不同物理化学和生物化学性质。有利于更好地发现理化性质之间的隐藏模式,从而得到潜在的规律,并可以有效的降低特征维数。
5.本发明的具体步骤如下:
6.步骤1、输入数据,其中输入数据为蛋白质序列;
7.步骤2、每条肽序列数据都用p=r1r2......r
l
表示,给定一个氨基酸指数aaindex,将上述序列编码成p
e
=e1e2......e
l
;其中e
i
为氨基酸残基的r
i
的属性值。
8.步骤3、构造字典;
9.在字典构造阶段,分为以下3步:
10.(1)、肽序列编码:利用n
‑
gram模型将肽序列分解成单词,建立编码序列;
11.(2)、编码序列分割:将编码序列分成不同长度的片段,如长度为2的片段和长度为3的片段。
12.(3)、字典中的单词:对长度相同的片段采用c均值聚类,以聚类中心作为字典中词。
13.步骤4、样本字典表示;
14.在样本字典表示阶段,分为3步,其中前两步与构造字典过程的前两步相同。
15.(1)、肽序列编码;
16.(2)、编码序列分割;
17.(3)、参考字典中单词对样本特征向量构造:特征由基于欧氏距离的词频构成:
[0018][0019]
其中,l为单词长度,c
l
是字典中长度为l的单词数,f
il
是第i个单词的出现频率。
[0020]
本发明的有益效果:本发明解决了传统特征提取方法中基于氨基酸组成和位置方法会丢失氨基酸相互关系、信息不全的问题,同时解决了基于理化性质方法时间计算开销大,特征维度过多的问题,是一种比较适用于高维数据集同时保持较高准确率的方法。
附图说明
[0021]
图1为本发明的算法总流程图;
[0022]
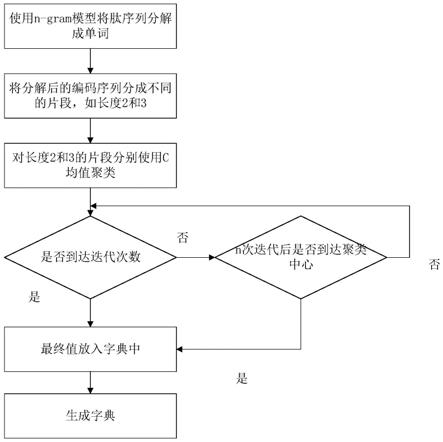
图2为本发明的字典生成流程图;
[0023]
图3为本发明生成特征矩阵流程图。
具体实施方式
[0024]
本实施例中没有详细说明的部分请参照发明内容的描述。
[0025]
如图1所示,一种基于字典学习的氨基酸序列特征提取方法,具体步骤如下:
[0026]
步骤1、输入数据集macppred,样本数量为532,其中非抗癌钛样本数量为266,正常样本数量为266。macppred可以从http://thegleelab.org/macppred/acpdata.html中下载获得。
[0027]
步骤2、利用氨基酸指数(aaindex)表示蛋白质序列:每条肽序列数据都可用p=r1r2......r
l
表示,给定一个aaindex,可将上述序列编码成p
e
=e1e2......e
l
。其中e
i
为氨基酸残基的r
i
的属性值。
[0028]
步骤3、构造字典,字典由aaindex数据库中八种理化性质聚类而成,具体流程图见图2。在字典生成阶段,分为以下三步:
[0029]
(1)肽序列编码:利用n
‑
gram模型将肽序列分解成单词,建立编码序列;
[0030]
(2)编码序列分割:将编码序列分成不同长度的片段,如长度为2的片段和长度为3的片段。
[0031]
(3)字典中的单词:对长度相同的片段采用c均值聚类,以聚类中心作为字典中词。
[0032]
步骤4、将样本集同样以氨基酸指数(aaindex)表示,并同样执行字典生成阶段的前两步,之后根据构造的字典生成特征向量,特征由基于欧几里得距离的词频构成。如公式(1):
[0033][0034]
其中,l为单词长度,c
l
是字典中长度为l的单词数,f
il
是第i个单词的出现频率。通过改变l的值,可以得到不同模式的特征,特别是当长度为1时,直接计算20个氨基酸出现的频率。生成特征矩阵流程图见图3。
[0035]
步骤5、通过支持向量机(support vector machine,简写svm)分类预测器进行分类。得到的性能参数如表1所示。表1为macppred(抗癌钛)数据集通过本发明得到并根据svm分类器分类的性能指标。
[0036]
accsnspmccauc0.9200.8920.9490.8420.968
[0037]
表1。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1