一种前列腺癌预后显著相关ceRNA调控网络的构建方法
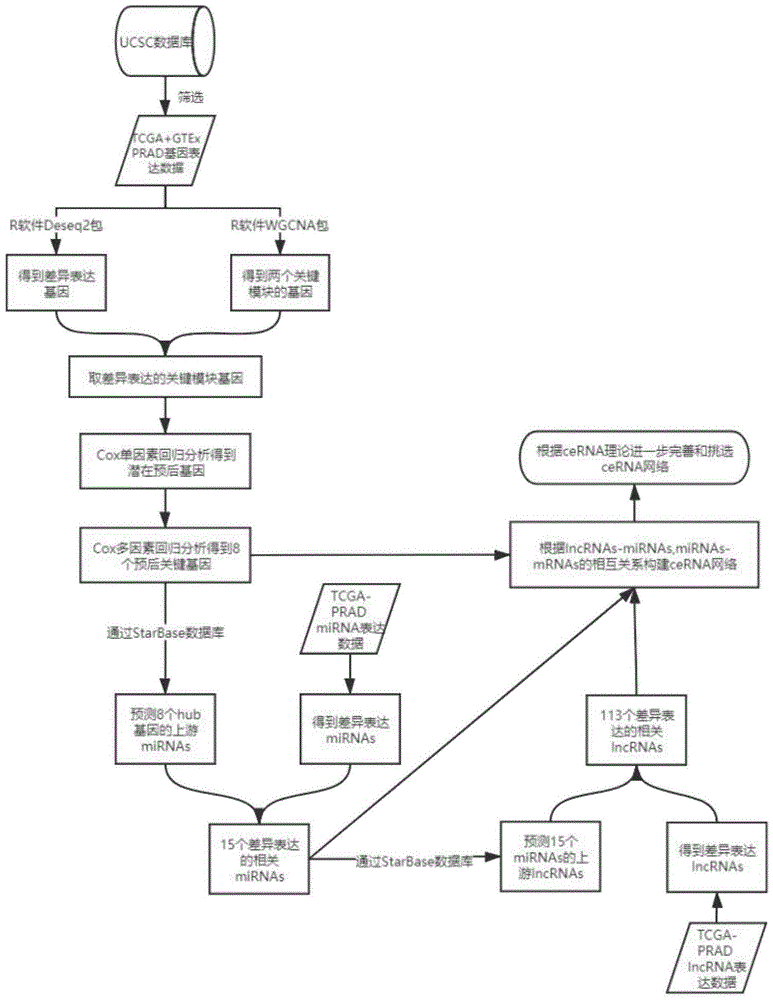
本发明具体涉及一种前列腺癌预后显著相关cerna调控网络的构建方法,属于医药
技术领域:
。
背景技术:
:前列腺癌是男性泌尿生殖系较为普遍的恶性肿瘤之一。在欧美等国家前列腺癌是男性发病率最高的恶性肿瘤,占癌症死亡率的第三位。从全球范围来看,发达国家的前列腺癌发病率高于发展中国家,目前美英法德日占据总发患者数的70%。我国的前列腺癌发病率虽低于欧美国家,但随着国家经济高速增长,近年来也呈明显上升趋势。目前,前列腺癌已成为中国男性泌尿生殖系肿瘤中排名第一位、所有肿瘤疾病中排名第五位的恶性肿瘤,是威胁老年男性健康的一个重要因素。众所周知,癌症的治疗一直都是现代医学领域难以攻克的难点,传统的治疗手段由于癌细胞随血液扩散导致很难彻底杀死癌细胞。研究前列腺癌生物学特性,寻求有价值的特异性生物标记物对前列腺癌早期诊断、靶向治疗和判断预后具有重要意义。探索生物标志物的潜在调控网络对于开发有效的治疗至关重要。近几年来,越来越多的证据揭示了mrna-mirna-lncrna调控网络在多种人类癌症中具有重要的调控作用。许多研究指出,cerna网络可能是前列腺癌预后的一个标志。因此利用mrna-mirna-lncrna相互作用关系为疾病的早期诊断,设计靶向药物进行精准治疗,对癌症的靶向治疗及个体化精准医疗有重要的意义。尽管,越来越多的证据揭示mrna-mirna-lncrna调控网络在多种人类癌症中发挥重要作用。然而,在癌症预后相关的mrna-mirna-lncrna调控网络研究仍然缺乏。技术实现要素:本发明的目的在于克服现有
背景技术:
中的不足,提供一种前列腺癌预后显著相关cerna调控网络的构建方法。为达到上述目的,本发明是采用下述技术方案实现的:提供一种前列腺癌预后显著相关cerna调控网络的构建方法,包括以下步骤:步骤1:从ucsc数据库中整合tcga和gtex前列腺癌归一化基因表达数据集,从tcga数据库中下载前列腺癌的基因表达数据及mrna、mirna表达数据、lncrna表达数据和前列腺癌病人的临床信息;步骤2:对tcga和gtex数据库前列腺癌归一化基因表达数据集进行差异基因表达分析,得到前列腺癌差异表达基因;步骤3:对tcga和gtex数据库前列腺癌归一化基因表达数据集进行加权基因共表达网络分析,将前列腺癌基因划分为高度相关的多个特征模块,并将特征模块与前列腺癌病人样本建立关联,找到发挥关键功能模块;取关键功能模块与前列腺癌差异表达基因的交集,得到差异表达的关键模块基因;步骤4:对关键模块基因进行cox单因素回归分析,得到潜在预后基因;对潜在预后基因进行cox多因素回归分析,筛选出关键基因;步骤5:根据关键基因预测得到关键基因的上游mirna,基于前列腺癌的mirna表达数据进行差异表达分析,筛选出差异表达的相关mirna;步骤6:根据差异表达的相关mirna进行预测,得到前列腺癌差异表达mirna的上游lncrna,基于前列腺癌的lncrna表达数据进行差异表达分析,筛选出差异表达的相关lncrna;步骤7:基于得到的关键基因、差异表达的相关mirna、差异表达的相关lncrna,根据其相互作用关系构建cerna网络;步骤8:基于cerna网络,并根据差异表达的相关lncrna、差异表达的相关mirna和关键基因在前列腺癌中的表达趋势进行评估,结合生存分析得到最终的前列腺癌预后显著相关的cerna调控网络。进一步的,所述步骤2中对tcga和gtex数据库前列腺癌归一化基因表达数据集,利用r软件deseq2包进行差异基因表达分析得到1组差异表达基因;差异表达基因分析的阈值条件设置为:|log2fc|>1.5,且fdr<0.05;fc为差异倍数,fdr伪发现率;满足阈值条件的基因为差异表达基因。进一步的,所述步骤3包括,基于tcga和gtex数据库前列腺癌归一化基因表达数据集,利用r软件wgcna包进行加权基因共表达网络分析,根据基因的共表达相似性模式,将基因划分为不同的聚类树,继续根据聚类树中所示模块间相似程度,定义高度阈值为0.25,将聚类树中高度低于该值的模块进一步合并,划分出几个更为明显的基因共表达模块;将基因共表达模块与tcga前列腺癌病人样本进行相关性分析,使用r软件中的cor函数计算共表达模块与前列腺癌病人之间的相关系数和相关系数的p值,得到p<0.05且相关系数排名靠前的两个共表达模块作为关键功能模块。进一步的,所述步骤4包括,使用tcga数据库中前列腺癌的基因表达数据和临床信息,利用r软件中survival包对差异表达的关键模块基因进行cox单因素回归分析,以wald检验p<0.05作为筛选标准,得到潜在的预后基因;使用tcga数据库中前列腺癌的基因表达数据和临床数据,利用r软件中survival包对预后基因进行cox多因素回归分析,对比多个cox模型的aic值,选取aic值最小的模型为最优模型,所述最优模型模型里的基因为关键基因。进一步的,所述步骤4还包括,基于关键基因根据以下公式计算预后风险评分,构建风险评估预后模型,其中n代表预后关键基因的数量,expi代表该基因的表达数据,ci代表通过cox多因素回归分析得到的关键基因估计回归系数。进一步的,所述步骤5包括:根据关键基因,利用starbase数据库预测关键基因的上游mirna;使用tcga数据库中前列腺癌的mirna表达数据,利用r软件中deseq2包,对前列腺癌的mirna表达数据进行差异表达分析,以|log2fc|>1,且fdr<0.05作为筛选标准,得到前列腺癌的差异表达mirna;取关键基因的上游mirna与前列腺癌的差异表达mirna的交集,得到关键基因的差异表达的相关mirna。进一步的,所述步骤6包括:对差异表达的相关mirna,利用starbase数据库进行预测得到前列腺癌差异表达的相关mirna的上游lncrna;利用r软件中deseq2包,对前列腺癌的lncrna表达数据进行差异表达分析,以|log2fc|>1,且fdr<0.05作为筛选标准,得到前列腺癌的差异表达lncrna;取差异表达的相关mirna的上游lncrna与前列腺癌的差异表达lncrna的交集,得到差异表达的相关lncrna。进一步的,所述步骤7包括:根据lncrna-mirna、mirna-mrna生物学关系,构建前列腺癌的潜在预后lncrna-mirna-mrna的cerna网络。进一步的,所述关键基因、差异表达的相关mirna、lncrna和lncrna-mirna、mirna-mrna相互作用关系分别导入cytoscape软件,使用cytoscape软件的可视化功能对构建的cerna网络进行可视化处理。进一步的,所述步骤8包括,利用r软件中survival和survminer包,使用kaplan-meier法,对差异表达的相关lncrna、差异表达的相关mirna和关键基因进行生存分析,以时序检验logrankp<0.05作为筛选标准,得到与前列腺癌预后显著相关的mrna、mirna和lncrna,结合表达趋势进行筛选得到最终的前列腺癌预后显著相关cerna调控网络。与现有技术相比,本发明所达到的有益效果:本发明提出的一种前列腺癌预后显著相关cerna调控网络的构建方法,基于高通量测序数据通过生物信息学进行整合分析筛选,对tcga和gtex表达数据集进行整合归一化后再进行差异表达分析,解决样本数量不平衡,便于挖掘稳健、准确的潜在生物学标志物;本发明通过wgcna分析和cox比例风险回归分析筛选关键基因,能够描述多个基因和生存之间的关系,使得相关系数分布更加符合无尺度网络分析,调控精确,对疾病早期诊断,设计靶向药物精准治疗具有重要作用。附图说明图1为本发明实施例一种前列腺癌预后显著相关cerna调控网络的构建方法流程图;图2是本发明实施例根据前列腺癌预后显著相关lncrna、mirna和mrna相互作用关系构建的cerna网络。具体实施方式下面结合附图对本发明作进一步描述。以下实施例仅用于更加清楚地说明本发明的技术方案,而不能以此来限制本发明的保护范围。如图1所示,为本发明实施例一种前列腺癌预后显著相关cerna调控网络的构建方法流程图,包括如下步骤:步骤1)从ucsc数据库中下载tcga和gtex前列腺癌归一化基因表达数据集,下载tcga(thecancergenomeatlas)数据库中前列腺癌的基因表达数据及mrna、mirna表达数据、lncrna表达数据和前列腺癌的临床信息;步骤2)对tcga和gtex数据库前列腺癌归一化基因表达数据进行差异基因表达分析,得到前列腺癌差异表达基因;步骤3)对tcga和gtex数据库前列腺癌归一化基因表达数据进行加权基因共表达网络分析(wgcna),将前列腺癌基因划分为高度相关的几个特征模块,并将模块与前列腺癌病人样本建立关联,从而找到发挥关键功能的基因;取关键模块的基因与步骤2)得到的差异表达基因作交集,得到差异表达的关键模块基因;步骤4)根据步骤3)得到的差异表达的关键模块基因,进行cox单因素回归分析,得到潜在预后基因;对潜在预后基因进一步进行cox多因素回归分析,筛选出关键基因(hubgene),并由关键基因(hubgene)构建风险评估预后模型;步骤5)根据步骤4)得到的关键基因(hubgene),进行预测得到关键基因(hubgene)的上游mirna,基于tcga-prad的mirna表达数据进行差异表达分析,筛选出差异表达的相关mirna;步骤6)根据步骤5)得到的mirna,进行预测得到前列腺癌差异表达mirna的上游lncrna,基于tcga-prad的lncrna表达数据进行差异表达分析,筛选出差异表达的相关lncrna;步骤7)基于得到的关键基因(hubgene)、差异表达的相关mirna、lncrna,根据其相互作用关系构建cerna网络;步骤8)基于步骤7)得到的cerna网络,并根据差异表达的相关lncrna、mirna和关键基因(hubgene)在前列腺癌中的表达趋势进行评估,结合生存分析得到最终的前列腺癌预后显著相关的cerna调控网络。图2是本发明实施例根据前列腺癌预后显著相关lncrna、mirna和mrna相互作用关系构建的cerna网络。在实施例中,前列腺癌预后显著相关cerna调控网络的构建方法,步骤2)中,对tcga和gtex数据库前列腺癌归一化基因表达数据,利用r软件deseq2包进行差异基因表达分析得到1组差异表达基因;上述差异基因表达分析的阈值条件设置为:|log2fc|>1.5,且fdr<0.05;fc为差异倍数,fdr伪发现率;满足阈值条件的基因为差异表达基因。在实施例中,前列腺癌预后显著相关cerna调控网络的构建方法,步骤3)中,对步骤2)得到的差异表达基因进行基因富集分析,得到基因富集分析结果;包括:利用david在线分析数据库,对差异表达基因进行go和kegg通路富集分析,得到差异表达基因在不同信号通道中的分类信息;并下载富集分析结果,取fdr<0.05的数据为有效数据。实施例中,前列腺癌预后显著相关cerna调控网络的构建方法,步骤3)中,对tcga和gtex数据库前列腺癌归一化基因表达数据进行加权基因共表达网络分析(wgcna),将前列腺癌基因划分为高度相关的几个特征模块,并将模块与前列腺癌病人样本建立关联,从而找到发挥关键功能的基因。取关键模块的基因与步骤2)得到的差异表达基因作交集,得到差异表达的关键模块基因,包括:基于tcga和gtex数据库前列腺癌归一化基因表达数据,利用r软件wgcna包进行加权基因共表达网络分析(wgcna),根据基因的共表达相似性模式,将基因划分为不同的聚类树,继续根据聚类树中所示模块间相似程度,定义高度阈值为0.25,将聚类树中高度低于该值的模块进一步合并,划分出几个更为明显的共表达模块。将通过加权基因共表达网络分析(wgcna)得到的基因共表达模块与tcga前列腺癌病人样本进行相关性分析,使用r软件中的cor函数计算共表达模块与前列腺癌病人之间的相关系数和相关系数的p值,得到p<0.05且相关系数排名靠前的两个共表达模块作为关键模块,把两个关键模块内的基因与步骤2)得到的差异表达基因取交集,得到差异表达的关键模块基因。实施例中,前列腺癌预后显著相关cerna调控网络的构建方法,步骤4)根据步骤3)得到的差异表达的关键模块基因,进行cox单因素回归分析,得到潜在预后基因;对潜在预后基因进一步进行cox多因素回归分析,筛选出关键基因(hubgene)。由关键基因构建风险评估预后模型,包括:使用tcga数据库中前列腺癌的基因表达数据和临床数据,利用r软件中survival包对差异表达的关键模块基因进行cox单因素回归分析,以wald检验(waldtest)p<0.05作为筛选标准,得到潜在的预后基因(如表1所示)。表1:关键模块基因进行cox单因素回归分析得到的潜在预后基因genebetahr(95%ci)wald.testpvaluepkmyt10.58651.7976(1.344-2.4044)15.620.0001tk10.06221.0642(1.0307-1.0988)14.520.0001plagl10.55721.7457(1.1778-2.5875)7.70.0055nlgn20.31671.3726(1.0563-1.7836)5.610.0178rpl22l10.02611.0265(1.0043-1.0491)5.480.0193nme20.05581.0574(1.0088-1.1084)5.40.0201tmem132a0.06961.0721(1.0071-1.1412)4.760.0291slc12a80.32031.3775(1.019-1.8623)4.340.0373dbndd10.13661.1464(1.0054-1.3072)4.160.0414pycr10.0251.0253(1.0002-1.051)3.910.0479使用tcga数据库中前列腺癌的基因表达数据和临床数据,利用r软件中survival包对上述得到的潜在预后基因进行cox多因素回归分析,对比多个cox模型的aic值(akaike信息准则),选取aic值最小的模型为最优模型,模型里的基因为关键基因(hubgene)(如表2所示)。表2:prad中的hubgene根据以下公式计算基于关键基因(hubgene)的预后风险评分(riskscore),构建风险评估预后模型。其中n代表预后关键基因的数量,expi代表该基因的表达数据,ci代表通过cox多因素回归分析得到的关键基因(hubgene)估计回归系数。预后风险评分为:riskscore=plagl1*0.32859+dbndd1*0.11770+pkmyt1*(-0.30405)+nme2*0.02629+tk1*0.03934+rpl22l1*0.02846+nlgn2*0.40079+slc12a8*0.31790。实施例中,前列腺癌预后显著相关cerna调控网络的构建方法,步骤5)根据步骤4)得到的关键基因(hubgene),进行预测得到关键基因(hubgene)的上游mirna,基于tcga-prad的mirna表达数据进行差异表达分析,筛选出差异表达的相关mirna,包括:利用starbase数据库预测关键基因的上游mirna;使用tcga数据库中前列腺癌的mirna表达数据,利用r软件中deseq2包,对mirna进行差异表达分析,以|log2fc|>1,且fdr<0.05作为筛选标准,得到前列腺癌的差异表达mirna;取关键基因的上游mirna与前列腺癌的差异表达mirna的交集,得到差异表达的关键基因上游mirna(如表3所示)。表3:prad中差异表达的相关mirna实施例中,前列腺癌预后显著相关cerna调控网络的构建方法,步骤6)根据步骤5)得到的mirna,进行预测得到前列腺癌差异表达mirna的上游lncrna,基于tcga-prad的lncrna表达数据进行差异表达分析,筛选出差异表达的相关lncrna,包括:利用starbase数据库预测得到差异表达的相关mirna的上游lncrna;使用tcga数据库中前列腺癌的lncrna表达数据,利用r软件中deseq2包,对lncrna进行差异表达分析,以|log2fc|>1,且fdr<0.05作为筛选标准,得到前列腺癌的差异表达lncrna;取差异表达的相关mirna的上游lncrna与前列腺癌的差异表达lncrna的交集,得到差异表达的相关lncrna(如表4所示)。表4:prad中差异表达的相关lncrna实施例中,步骤7)基于得到的关键基因(hubgene)、差异表达的相关mirna、lncrna,根据其相互作用关系构建cerna网络,包括:基于得到的关键基因(hubgene)、差异表达的相关mirna、lncrna,根据lncrna-mirna、mirna-mrna相互作用关系,构建得到前列腺癌潜在预后lncrna-mirna-mrna的cerna网络;进一步的,将关键基因(hubgene)、差异表达的相关mirna、lncrna和lncrna-mirna、mirna-mrna相互作用关系分别导入cytoscape软件,使用cytoscape软件的可视化功能对构建的cerna网络进行可视化。实施例中,前列腺癌预后显著相关cerna调控网络的构建方法,步骤8)基于步骤7)得到的潜在预后cerna网络,利用r软件中survival和survminer包,使用kaplan-meier法,对cerna网络中的关键基因(hubgene)、差异表达的相关mirna、lncrna进行生存分析,以时序检验logrankp<0.05作为筛选标准,得到与前列腺癌预后显著相关的关键基因(hubgene)、mirna和lncrna,并根据与前列腺癌预后显著相关的lncrna、mirna和关键基因(hubgene)在前列腺癌中的表达趋势进行评估,根据表达量差异进行筛选得到最终的前列腺癌预后显著相关cerna调控网络(如表5所示)。表5:构建的前列腺癌预后显著相关cerna调控网络lncrnamirnamrnasnhg3mir-222-3ptk1结合图2和表5可知:由8个关键基因(hubgene)构建的风险评估预后模型能够有效预测前列腺癌患者未来的生存概率,可以作为一项独立的预后因素,并且基于这8个关键基因(hubgene),15个差异表达的相关mirnas和113个差异表达的相关lncrnas构建了前列腺癌cerna网络。然后,进一步分析了这些基因在前列腺癌中的生存情况和表达模式,发现snhg3与tk1具有一致的表达模式(在前列腺癌中显著上调),并且两者都具有与中位mirnamir-222-3p相反的表达模式。因此,最终获得了tk1-mir-222-3p-snhg3作为前列腺癌的潜在预后标志物。它们在前列腺癌中都具有潜在的预后价值,并且tk1和snhg3具有一致的趋势。在实际应用中,例如用于基因深度分析,还包括对得到的共有的差异表达基因进行基因富集分析,得到基因富集分析结果;具体包括:利用david在线分析数据库,对共有的差异表达基因进行go和kegg通路富集分析,得到差异表达基因在不同信号通道中的分类信息;并下载富集分析结果,取fdr<0.05的数据为有效数据。以上所述仅是本发明的优选实施方式,应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变形,这些改进和变形也应视为本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种制备自噬小体型瘤苗的培养基添加剂及其制备方法与制造工艺
- N-萘烷基取代的含氮杂环衍生物、制备方法及其抗肿瘤用途与制造工艺
- 作为BET蛋白抑制剂的1H‑吡咯并[2,3‑c]吡啶‑7(6H)‑酮和吡唑并[3,4‑c]吡啶‑7(6H)‑酮的制造方法与工艺
- 一种人肿瘤抑制蛋白变体及其应用的制造方法与工艺
- 一种人肿瘤抑制蛋白变体及其应用的制造方法与工艺
- G蛋白信号调节因子6及其抑制剂在治疗脂肪肝和Ⅱ型糖尿病中的功能和应用的制造方法与工艺
- 蛋白酶体抑制剂在制备降低动物脂含量或脂储积能力产品中的应用的制造方法与工艺
- GINS2基因或蛋白的抑制剂在制备抗肿瘤药物中的应用的制造方法与工艺
- ZNF383蛋白在制备抑制p53蛋白活性的产品中的应用的制造方法与工艺
- 蛋白酶体抑制剂MLN9708的合成方法与制造工艺