一种pH敏感的纳米胶束及其制备方法和应用
本发明涉及药物载体技术领域,尤其是一种ph敏感的纳米胶束及其制备方法和应用。
背景技术:
系统治疗(包括化疗、分子靶向药物治疗)是肿瘤最主要的治疗手段之一,也常与其它方法联合应用以最大限度地杀灭肿瘤细胞,使患者生存获益。然而,系统治疗为全身用药,药物作用的组织或细胞选择性不强,在杀伤肿瘤的同时也不可避免地对正常组织产生损伤,导致明显的毒副反应。而且,大多数化疗药物或分子靶向药物为疏水性药物,溶解度差,有效半衰期短,生物利用率低。因此,如何提高药物的生物利用率,在增强疗效的同时降低毒副作用是肿瘤系统治疗所面临的重要难题。
近来,拥有独特“核-壳结构”的聚合物纳米胶束因其具有作为药物载体及影像探针的应用前景而倍受关注。纳米胶束的疏水性内核可用于负载疏水性药物,如索拉非尼(sor)、紫杉醇和阿霉素等;而亲水性外壳则使其具有可溶性,能逃逸网状内皮系统的诱捕,延长药物循环时间,提高药物溶解度和生物利用率,从而增强其治疗效果。此外,纳米药物递送体系还可通过高渗透滞留效应在肿瘤组织聚集,提高肿瘤局部的药物浓度。而且,以纳米胶束制备的纳米药物还能进行表面功能化修饰,赋予其主动靶向特定组织或细胞的功能,进一步提高细胞对纳米药物的摄取效率及纳米药物在肿瘤组织内的聚集,当药物从纳米胶束中释放后,即可起到高效的抗肿瘤作用。
然而,就算纳米载体能高效地将药物输送至肿瘤组织,如果其负载的药物无法快速释放,达到足够的游离浓度以杀伤肿瘤细胞,其治疗效果也将大打折扣,甚至引起耐药,导致治疗失败。而许多传统纳米载体的药物释放特性就表现为:在制备后初期,其在储存或血液循环时会出现一过性的药物快速释放,但剩余的药物则常常需要持续几天甚至更长的时间来释放。如此,早期释放的药物将可能导致毒副反应的发生,而后期药物释放缓慢,就算纳米药物聚集于肿瘤组织,也无法达到杀伤肿瘤细胞的游离药物浓度,无法取得满意的治疗效果。
技术实现要素:
本发明的目的在于克服上述现有技术的不足,提供一种ph敏感的纳米胶束,其可在肿瘤组织聚集,提高肿瘤组织药物浓度;同时,该纳米胶束具有ph敏感的药物释放特性,可在肿瘤(细胞)弱酸性环境的触发下快速释放负载的药物;并且,该纳米胶束还可进行表面功能化修饰,获得靶向特定细胞的功能;再者,该纳米胶束负载磁共振成像(mri)造影剂超顺磁性氧化铁颗粒(spion)后可成为影像探针,以此实现肿瘤组织聚集、定点智能药物释放和肿瘤成像的诊疗一体化功能。
为实现上述目的,本发明采取的技术方案包括以下几个方面:
在第一个方面,本发明提供了一种ph敏感的纳米胶束,所述纳米胶束是由亲水性嵌段和疏水性ph敏感嵌段自组装形成的三嵌段聚合物,所述亲水性嵌段为聚乙二醇(peg);所述疏水性ph敏感嵌段为聚天冬氨酸(二异丙基乙二胺)-聚天冬氨酸(二正丁基乙二胺)(pasp(dip)-pasp(dba))。
所述亲水性嵌段为聚乙二醇(peg),其分子量为2kda;所述疏水性ph敏感嵌段为聚天冬氨酸(二异丙基乙二胺)-聚天冬氨酸(二正丁基乙二胺)(pasp(dip)-pasp(dba))。其中,pasp(dip)-pasp(dba)作为纳米胶束的疏水链段,能在弱酸性环境中质子化而转化为亲水链段,负载的疏水性药物则随着释放;而引入的亲水链段peg,则是为了增加纳米胶束的稳定性及生物相容性。
ph敏感的药物释放特性通过疏水性ph敏感嵌段来实现,这种微环境响应的药物递送体系在生理环境中药物释放缓慢,而在肿瘤细胞内溶酶体弱酸性环境的触发下则可快速地释放药物,从而显著增强药物的作用效果。
特别说明的是,本发明的纳米胶束除了可用于负载药物外,还可以用于负载其它疏水性化合物。
本发明的纳米胶束可增加疏水性药物的溶解度,延长药物循环时间,增加肿瘤组织的药物浓度,整合微环境响应的嵌段可使其在肿瘤组织内快速释放药物,提高游离药物浓度,以增强抗肿瘤效果。并且,通过表面的功能化修饰,可使其具有靶向特定细胞的功能,能进一步增加细胞对纳米药物的摄取及肿瘤组织的药物聚集。同时,该纳米胶束在负载spion后还具备mri探针的功能,使其成为诊疗一体化的纳米药物体系。
在第二个方面,本发明提供了上述ph敏感纳米胶束的制备方法,包括以下步骤:
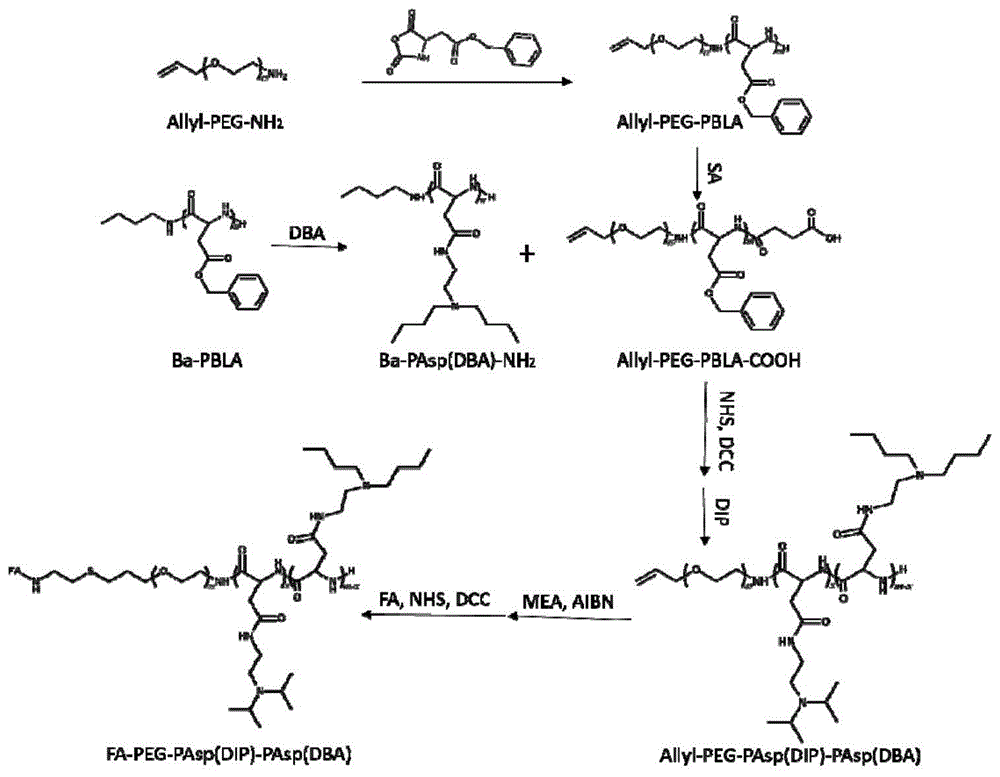
s1、将萘置于反应瓶内,在氩气保护下加入新蒸四氢呋喃(thf)溶解,再加入金属钾制成萘钾试剂;取烯丙基聚乙二醇羟基(allyl-peg-oh;分子量优选为2kda)溶解于新蒸苯溶剂,在氩气保护下滴加入上述萘钾试剂至溶液成墨绿色且不褪色,搅拌,加入对甲苯磺酰氯(tscl),搅拌,旋蒸浓缩,沉淀于冷乙醚,抽滤,干燥,得到黄色固体粗产品;
s2、将s1所得的黄色固体加入氨水内,搅拌、旋蒸浓缩,加入过量氯化钠(nacl)至饱和,用氯仿(chcl3)萃取,加入稀盐酸(hcl)搅拌;萃取,chcl3层加入氢氧化钠(naoh)溶液搅拌;萃取,chcl3层再加入碳酸氢钠(nahco3)溶液搅拌,萃取;萃取液中加入过量无水硫酸钠(na2so4)干燥,过滤,浓缩,沉淀,抽滤,干燥,获得烯丙基聚乙二醇氨基(allyl-peg-nh2);
s3、以s2所得allyl-peg-nh2为引发剂,在二氯甲烷与无水n,n-二甲基甲酰胺(dmf)的混合溶剂中引发苄氧羰基天冬氨酸酸酐(bla-nca)开环聚合,得到烯丙基聚乙二醇-聚天冬氨酸苄酯(allyl-peg-pbla),其分子量为6.1kda;
s4、将s3所得的allyl-peg-pbla和丁二酸酐溶解于chcl3中,70℃搅拌后沉淀于无水乙醇中,分离,分别用无水乙醇和双蒸水洗涤,冻干后得到羧酸化的allyl-peg-pbla-cooh;
s5、在氮气保护下将正丁胺(ba)溶解于二氯甲烷中,将bla-nca溶解于dmf,滴入ba溶液,搅拌后沉淀于无水冷乙醚,乙醚洗涤,干燥,得到ba-pbla,其分子量为7.18kda;
s6、取s5所得的ba-pbla溶解于无水二甲亚砜(dmso),加入n,n-二正丁基乙二胺(dba)进行氨解,于甲醇中透析,得到聚天冬氨酸(二正丁基乙二胺)氨基(pasp(dba)-nh2),其分子量为9.42kda;
s7、在氮气保护下,将s4所得的allyl-peg-pbla-cooh和n-羟基丁二酰亚胺(nhs)、n,n-二环己基碳酰亚胺(dcc)溶解于chcl3中,搅拌后加入s6所得的pasp(dba)-nh2,反应后过滤,沉淀于乙醚,依次经无水乙醇、双蒸水、无水乙醇及乙醚洗涤,干燥后得到allyl-peg-pbla-pasp(dba),其分子量为15.52kda;
s8、在氮气保护下将s7所得的allyl-peg-pbla-pasp(dba)溶解于dmso中,加入n,n-二异丙基乙二胺(dip)进行氨解,透析,冻干后得到三嵌段聚合物allyl-peg-pasp(dip)-pasp(dba),其分子量为16.25kda。
n,n-二异丙基乙二胺的pka≈8.91,而n,n-二正丁胺基乙二胺的pka≈6.72。在纳米胶束的合成过程中,如果只是单纯地使用的二异丙基乙二胺进行氨解,则合成胶束的稳定性不足,在生理环境中(ph7.4)即会出现解体,使药物提前释放;如果只是单纯地使用二正丁胺基乙二胺进行氨解,则合成的胶束过于稳固,ph敏感性不足,即使是在酸性条件下也难以有效释放药物。因此,本发明首先使用二正丁胺基乙二胺对正丁胺-聚天冬氨酸苄酯进行氨解,获得聚天冬氨酸(二正丁胺基乙二胺)氨基;然后,由聚天冬氨酸(二正丁胺基乙二胺)氨基和烯丙基聚乙二醇-聚天冬氨酸苄酯羧酸合成烯丙基聚乙二醇-聚天冬氨酸苄酯-聚天冬氨酸(二正丁胺基乙二胺);再使用二异丙基乙二胺进行氨解,制备三嵌段聚合物烯丙基聚乙二醇-聚天冬氨酸(二异丙基乙二胺)-聚天冬氨酸(二正丁基乙二胺)。该聚合物中聚天冬氨酸(二异丙基乙二胺)和聚天冬氨酸(二正丁基乙二胺)两嵌段可调节聚合物整体的pka,使聚合物具有适当的ph敏感性,在生理环境中能稳定地负载药物,而在肿瘤细胞酸性溶酶体内则能快速地释放药物。
在第三个方面,本发明提供了采用第二个方面的方法制备的ph敏感纳米胶束。
在第四个方面,本发明提供了第一个方面或第三个方面的纳米胶束在制备肿瘤靶向纳米药物中的应用。
在第五个方面,本发明提供了一种肿瘤靶向纳米药物,所述肿瘤靶向纳米药物由载体和负载的制剂组成,所述载体为第三个方面的ph敏感纳米胶束,所述负载的制剂为抗肿瘤药物和/或mri造影剂spion。
需要说明的是,所述的肿瘤靶向纳米药物可延长药物的循环时间,增加药物在肿瘤组织的聚集和渗透性;而且,该纳米药物具有肿瘤细胞靶向性,具有主动靶向的功能;再者,该纳米药物具有肿瘤细胞微环境响应性ph敏感药物释放特性,在其进入肿瘤细胞后可在溶酶体弱酸性环境的触发下快速释放负载的药物;另外,当该纳米胶束负载spion后,便具备了mri探针的功能,能用于肿瘤成像和药物分布情况的监测。这种利用纳米药物的靶向肿瘤聚集、微环境响应释药来提高肿瘤治疗效果,同时赋予纳米药物mri可视化功能的方法,为肿瘤的诊疗提供了一种创新性的策略。本发明的纳米药物中,负载的抗肿瘤药物包括但不限于索拉非尼(sor)、阿霉素、紫杉醇、甲氨蝶呤、拓扑替康等。
优选地,所述肿瘤靶向纳米药物的粒径为100nm,能增加纳米药物在肿瘤组织的聚集和渗透性,所述肿瘤靶向纳米药物的表面ζ电位为24mv。
在第六个方面,本发明提供了一种肿瘤靶向纳米药物的制备方法,所述制备方法包括如下步骤:
s9、取allyl-peg-pasp(dip)-pasp(dba)、巯基乙胺(mea)和偶氮二异丁晴(aibn)溶解于dmso中,搅拌,透析,得到nh2-peg-pasp(dip)-pasp(dba);
s10、于dmso中将叶酸(fa)连接至s9所得nh2-peg-pasp(dip)-pasp(dba)的peg端氨基上,过滤后于氢氧化钾(koh)溶解中透析,chcl3多次萃取,无水硫酸镁(mgso4)干燥后得到fa修饰的三嵌段聚合物fa-peg-pasp(dip)-pasp(dba);
s11、用dmso溶解s10所得的fa-peg-pasp(dip)-pasp(dba)和抗肿瘤药物,用氯仿溶解spion,将溶液混合后采用超声乳化法制得磁共振可视化的靶向纳米药物fppass。
优选地,步骤s11中,聚合物、抗肿瘤药物和spion的投料质量比优选为10:1:1。
另外,在合成过程中若使用单甲氧基聚乙二醇羟基(mpeg-oh)代替allyl-peg-oh(不使用fa修饰),则最终可合成非靶向的纳米药物ppass。
在上述肿瘤靶向纳米药物的制备方法中,抗肿瘤药物包括但不限于索拉非尼(sor)、阿霉素、紫杉醇、甲氨蝶呤、拓扑替康等。
综上所述,本发明的有益效果是:
本发明的ph敏感的纳米胶束可在肿瘤部位聚集,增加肿瘤组织药物浓度;同时,该纳米胶束具有微环境触发的药物释放特性,在生理环境(ph7.4)中药物逃逸极少,可避免游离药物对正常组织的损伤,而在肿瘤细胞溶酶体内弱酸性环境(ph约为5.0)中则药物可快速释放,游离药物浓度升高,其抗肿瘤效果增强;另外,该纳米胶束还可以通过表面的功能化修饰使其具有靶向特定细胞、肿瘤组织的功能;再者,该纳米胶束在负载spion后便具备了mri探针的功能,可用于肿瘤成像和药物体内分布的监测。
附图说明
图1为本发明中的ph敏感的纳米胶束的合成线路图;
图2为实施例3叶酸修饰后聚合物(纳米胶束)的1h磁共振波谱分析结果;
图3为实施例3制备的肿瘤靶向纳米药物的透视电子显微镜照片;
图4为实施例3制备的肿瘤靶向纳米药物在ph7.4和ph5.0条件下的药物释放曲线;
图5a为肝癌bel-7402细胞对纳米药物摄取情况的流式细胞术检测结果;图5b为各组细胞流式细胞术检测的平均荧光强度;
图6a为纳米药物处理后bel-7402细胞样品的mri扫描t2wi图像;图6b为细胞样品t2wi图像上t2信号的变化情况;图6c为纳米药物处理后bel-7402细胞样品t2-map图像;图6d为细胞样品t2-map图像上t2值的变化情况;
图7a为未负载药物的靶向/非靶向纳米胶束fppas和ppas的细胞毒性试验结果;图7b为游离sor、靶向纳米药物fppass和非靶向纳米药物ppass的细胞毒性试验结果。
具体实施方式
为更好的说明本发明的目的、技术方案和优点,下面将结合附图和具体实施例对本发明作进一步说明。如无特别说明,本申请中的试剂浓度均采用质量浓度。
实施例1
本发明中ph敏感的纳米胶束的一种实施例,本实施例的纳米胶束是由亲水性嵌段和疏水嵌段的三嵌段聚合物自组装形成;其中,亲水性嵌段为peg,疏水性嵌段为pasp(dip)-pasp(dba);peg的分子量优选为2kda,最终三嵌段聚合物peg-pasp(dip)-pasp(dba)的分子量为16.25kda。
本发明中肿瘤靶向纳米药物的一种实施例,由fa修饰后的纳米胶束fa-peg-pasp(dip)-pasp(dba)负载sor和spion后制成;该靶向纳米药物的粒径约为100nm,表面ζ电位约为24mv。
本发明中肿瘤靶向纳米药物制备方法的一种实施例,包括如下步骤:采用超声乳化法诱导三嵌段聚合物fa-peg-pasp(dip)-pasp(dba)、mri造影剂spion和抗肿瘤药物sor自组装制得mri可视化的靶向纳米药物fppass,其中聚合物、spion与sor的投料质量比优选为10:1:1。
实施例2ph敏感的纳米胶束的合成
本发明的ph敏感的纳米胶束制备方法的一种实施例,本发明的纳米胶束的合成路线如图1所示,具体包括如下步骤:
首先,以allyl-peg-oh为原料合成allyl-peg--nh2,具体地,称取1.03g萘(8mmol)置于100ml反应瓶内,干燥后在氩气保护下加入30ml新蒸thf溶解,加入金属钾溶解后制成萘钾试剂;称取10gallyl-peg-oh置于250ml双口瓶中,70℃真空下干燥,恢复至室温后加入120ml新蒸苯溶剂;在氩气保护下将上述萘钾试剂缓慢滴加至allyl-peg-oh苯溶液中,至溶液变成墨绿色且不褪色后停止滴加,室温下搅拌30分钟,加入1.22gtscl(6.4mmol),搅拌6小时;反应液旋蒸浓缩至30ml,沉淀于10倍量以上的冷乙醚,抽滤,干燥,得到黄色固体;然后,将黄色固体溶解于大量的氨水(23%~28%)中,密封,室温下搅拌至少5天,旋蒸浓缩至100ml以内,加入过量nacl至溶液饱和,用chcl3萃取,加入120mlhcl(3mol/l)搅拌2小时,萃取;chcl3层加入naoh溶液(6mol/l)搅拌2小时以上,萃取;加入120mlnahco3搅拌2小时以上,萃取;萃取液加入过量无水na2so4干燥1小时以上,过滤,浓缩,500ml无水乙醚中沉淀,抽滤,真空干燥,得到allyl-peg-nh2;
把allyl-peg-nh2作为大分子引发剂引发bla-nca聚合:取0.5gallyl-peg--nh2(0.25mmol)置于50ml反应瓶内,70℃真空干燥2小时,冷却至室温,在氮气保护下加入新蒸二氯甲烷20ml溶解,并加入用2mldmf溶解的1.25gbla-nca(5mmol),35℃搅拌48小时,将反应液沉淀于300ml冷乙醚中,抽滤,用乙醚洗涤3次,真空干燥后得到allyl-peg-pbla;
allyl-peg-pbla的羧酸化:取0.14g丁二酸酐(1.4mmol)和0.85gallyl-peg-pbla(0.14mmol)溶解于chcl3中,70℃持续搅拌反应48小时,反应液沉淀于无水冷乙醇,分流后无水乙醇和双蒸水分别洗涤3次,冻干后等到allyl-peg-pbla-cooh;
制备ba-pbla:在氮气保护下将37.2μl新蒸ba(0.74g/ml,0.37mmol)加入至40ml新蒸二氯甲烷中;取3.23gbla-nca(12.95mmol)溶解于dmf中,并加入至ba溶液内,35℃搅拌48小时以上;将反应液沉淀于500ml冷乙醚,洗涤,真空干燥,获得ba-pbla;
用dba与ba-pbla进行氨解反应:取2gba-pbla(0.28mmol)置于50ml反应瓶内,在氮气保护下加入20mldmso,加入3.4gdba(19.6mmol),35℃搅拌反应12小时;于无水甲醇透析(截留分子量1kda)2天,旋蒸,干燥,得到pasp(dba)-nh2;
通过酰胺化反应合成allyl-peg-pbla-pasp(dba):在氮气保护下,于100ml反应瓶中加入1.5gallyl-peg-pbla-cooh(0.25mmol)、57mgnhs(0.5mmol)和96mgdcc(0.5mmol),加入40ml新蒸chcl3溶解,室温搅拌4小时;加入2.4gpasp(dba)-nh2(0.25mmol),室温下反应24小时,过滤除去不溶物二环己基脲(dcu),将溶液沉淀于乙醚,过滤,依次经无水乙醇、ph5.0双蒸水、无水乙醇和乙醚洗涤,干燥后得到allyl-peg-pbla-pasp(dba);
使用dip进行氨解:在氮气保护下,于50ml反应瓶内加入1.55gallyl-peg-pbla-pasp(dba)(0.1mmol)和0.58gdip(4mmol),加入15mldmso溶解,35℃搅拌12小时;反应液于双蒸水中透析(截留分子量3.5kda)2天,冻干后得到allyl-peg-pasp(dip)-pasp(dba)。
实施例3肿瘤靶向纳米药物的制备及其理化性质
取实施例2制备的allyl-peg-pasp(dip)-pasp(dba)1.63g(0.1mmol)、mea78mg(1mmol)和aibn2.5mg(0.015mmol)置于50ml反应瓶内,加入dmso溶解,70℃搅拌48小时;反应液于双蒸水中透析48小时,冻干后得到nh2-peg-pasp(dip)-pasp(dba);
采用类似于合成allyl-peg-pbla-pasp(dba)步骤中的酰胺化反应方法将fa连接至nh2-peg-pasp(dip)-pasp(dba)上peg端的氨基上,过滤去除dcu;将反应液置于koh溶液(ph9~ph10)中透析48小时,后用chcl3多次萃取,无水mgso4干燥,旋干后获得fa修饰的聚合物fa-peg-pasp(dip)-pasp(dba);
将4mgspion溶解于1.5mlchcl3中;将40mgfa-peg-pasp(dip)-pasp(dba)和4mgsor溶解于0.5mldmso中;将二种溶液混合均匀,取20ml磷酸盐缓冲液(pbs)加入小烧杯内,冰水浴中在超声发生器的作用下将上述混合溶液缓慢滴入pbs中进行乳化,旋蒸至液体变澄清,于pbs中透析(截留分子量14kda)24小时,过滤去除大颗粒,超滤,浓缩至4ml,得到靶向纳米药物fppass。
上述纳米药物制备过程中,若使用mpeg-oh代替allyl-peg-oh(不使用fa进行靶向修饰),则最终可制备非靶向纳米药物ppass。制备过程中若不添加药物sor,则可获得负载spion的未载药的靶向纳米胶束fppas和非靶向纳米胶束ppas。
1h磁共振波谱分析结果(图2)示:6.60、7.55和8.55ppm处出现了fa质子的特征位移峰;1.05ppm处出现dip上甲基氢((ch3)2ch-)的特征峰(图2a);0.79和1.15ppm处分别为dba上甲基氢(ch3ch2-)和亚甲基氢(ch3(ch2)2-ch2-)的特征峰(图2b和c);3.5ppm处出现peg亚甲基氢(-och2ch2-)的特征峰(图2d)。这说明了聚合物中含有各嵌段的成分,fa-peg-pasp(dip)-pasp(dba)合成成功。
粒径和电位检查结果显示,纳米胶束的粒径分别为ppas96.2±3.2nm、fppas98.2±3.7nm、ppass99.7±3.9nm和fppass101.4±4.6nm;表面电位分别为ppas24.23±2.23mv、fppas23.28±2.23mv、ppass24.42±2.59mv和fppass23.79±1.91mv。透视电镜照片显示,靶向纳米药物fppass为均匀的球形结构,直径约为100nm(图3)。
纳米药物的体外药物释放试验的结果如图4所示,靶向纳米药物fppass在ph7.4环境中药物释放缓慢,48小时的药物释放率仅为12.6%;而在ph5.0条件下,纳米胶束释药效率明显增快,6小时达49%,24小时已接近80%。这验证了该纳米胶束具有ph敏感的药物释放特性。
实施例4细胞摄取能力的检测
发明人采用流式细胞仪来检测实施例3的纳米药物被bel-7402细胞摄取的效率。用疏水性荧光染料尼罗红代替sor进行药物的包封,制备荧光标记的靶向纳米药物和非靶向纳米药物。将靶向、非靶向纳米药物与bel-7402细胞孵育2小时后消化、重悬细胞,于流式细胞仪上检测尼罗红阳性细胞的比例及荧光强度。同时,进行游离配体竞争抑制试验(即在靶向纳米药物与细胞孵育前用fa与细胞孵育4小时进行竞争性抑制),验证fa介导增强了肿瘤细胞对纳米药物的摄取。
结果如图5a、b所示,bel-7402细胞与靶向纳米药物孵育后,细胞的平均荧光强度明显高于非靶向纳米药物组及竞争抑制组的细胞荧光强度。这说明了fa修饰的靶向纳米药物具有肿瘤细胞靶向性,更易于被bel-7402细胞摄取,更利于药物的递送。
实施例5纳米药物的成像潜力检测
发明人采用细胞mri扫描来评估实施例3纳米药物的mri显像潜能。将bel-7402细胞种于6孔板内(1.5×106个/孔),孵育过夜,加入不同浓度的靶向、非靶向纳米药物(实施例3中制备的fppass和ppass)孵育2小时,消化、收集细胞,重悬于400μl2%的琼脂糖溶液并添加至酶标板内,用3t磁共振扫描仪进行t2加权成像(t2wi)和t2-mapping成像。
结果如图6a、b、c和d所示,随着与细胞孵育纳米药物铁浓度的增加,细胞样品于t2wi和t2-map图像上的信号强度和t2值逐渐降低;而且,在相同的铁浓度下,相比较于非靶向纳米药物ppass,与靶向纳米药物fppass孵育后的细胞信号强度和t2值下降更为明显。这说明了bel-7402细胞经fppass处理后,细胞内含有更多的spion,引起了更为显著的t2信号下降。这些结果验证了制备的纳米药物具有良好的显像和示踪效能,也再次验证了fa修饰后的靶向纳米药物具有肿瘤靶向性(高表达叶酸受体的肿瘤细胞),能促进bel-7402细胞对它的摄取效率,将更有利于药物的输送。
实施例6纳米药物的抗肿瘤细胞效果评估
发明人通过cck-8细胞活性试验来评估纳米药物的抗肿瘤效用:bel-7402细胞(5×103个/孔)种于96孔板内,孵育过夜,加入不同浓度的实施例3中制备的纳米胶束fppas、ppas和fppass、ppass,孵育48小时,每孔加入cck-8溶液10μl,继续孵育3小时后于酶标仪检测450nm处的吸收值。
如图7a所示,非载药纳米胶束fppas、ppas的细胞毒性很低,当胶束浓度达720μg/ml时,细胞活性仍高于80%,说明本发明中用于负载药物的纳米胶束的生物相容性好。图7b显示负载sor后的纳米药物呈现出明显的细胞毒性作用,且靶向纳米药物fppass的毒性作用明显高于非靶向纳米药物ppass。
最后应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!