L-4-氯犬尿氨酸的剂型和治疗用途的制作方法
本申请是分案申请,其原案申请的申请号为201480010060.x(国际申请号为pct/us2014/012598)、申请日为2014年1月22日、发明名称为“l-4-氯犬尿氨酸的剂型和治疗用途”,申请日“2014年1月22日”为国际申请日。
本发明涉及单位剂型的l-4-氯犬尿氨酸(l-4-cl-kyn)的组合物以及这些组合物用于治疗神经疾病的用途。
背景技术:
谷氨酸是哺乳动物中枢神经系统中的主要的兴奋性神经递质,并且谷氨酸涉及若干不同路径的调节。过量的内源性谷氨酸和急性与慢性的各种神经障碍之间已经被报道存在关联,神经障碍诸如脑缺血、癫痫、肌萎缩性脊髓侧索硬化症、亨廷顿氏病、帕金森氏病和阿尔茨海默氏病。
已知通过n-甲基-d-天门冬氨酸(nmda)受体(nmda-r)的过度活跃的谷氨酸能突触传递在诸如神经性疼痛的若干神经疾病中起着关键作用。然而,直接作用nmda-r拮抗剂产生诸如精神病的许多副作用,这限制了它们的医疗用途。也可以通过阻塞nmda-r上的调节位点来实现bnda-r的拮抗作用,该调节位点称为甘氨酸b(glyb)协同激动剂位点。(参考文献8,下文呈现了对该参考文献的引证和以该方式引用的其他参考文献)。当与传统的nmda-r拮抗剂相比时,glyb拮抗剂具有好得多的安全状况并且不引起与“传统”的nmda-r拮抗剂关联的不利的副作用。(参考文献1、6和10)。
glyb拮抗剂也已经在体外和神经性疼痛动物模型中展示出减小痛觉过敏和触诱发痛,并且比传统的nmda-r拮抗剂具有更少的副作用,从而使它们成为用作潜在的镇痛药的更安全的替代物。例如,见参考文献2。
目前已知的一种最有效和特定的glyb拮抗剂是7-氯犬尿喹啉酸(7-cl-kyna),7-氯犬尿喹啉酸是内源性神经调质犬尿喹啉酸的合成的氯化类似物。7-氯犬尿喹啉酸已经展示出防止兴奋毒性和缺血性神经元损伤,但是像大多数glyb拮抗剂一样不能穿越血脑屏障。因此,限制了它的临床使用。(参考文献4和9)。
相反,7-氯犬尿喹啉酸的前药l-4-氯犬尿氨酸在给药之后容易进入中枢神经系统(cns)。(参考文献3、5、11和12)。l-4-氯犬尿氨酸在活化的星形胶质细胞内有效地转化为7-氯犬尿喹啉酸,(参考文献5),并且脑中的7-氯犬尿喹啉酸的水平由于星形胶质细胞活化而在神经元损伤或兴奋毒性损害的位点处增高。(参考文献5)。
在临床前的研究中,l-4-氯犬尿氨酸已经在大鼠中展示出抗癫痫活性。(参考文献11)。也发现该化合物增大放电率并且在大鼠的脑中爆发多巴胺能神经元的放电活动。(参考文献7)。
在美国专利第5,547,991号(1996)中palfreyman等人描述了用于合成一类4,6-双取代的犬尿氨酸衍生物(包括l-4-氯犬尿氨酸)的方法以及它们作为nmda受体的拮抗剂的用途。也描述了包含这些化合物的药物组合物和它们的治疗用途。
技术实现要素:
在形成本专利申请的部分的权利要求中陈述了本发明和各个实施例。
不限制以上所述,在优选的方面中,本发明涉及一种药物组合物,该药物组合物的每单位剂量基本上由约360mg、1080mg或1440mg的量的l-4-氯犬尿氨酸与诸如载体和赋形剂的药学上可接受的组分组成。本发明的另一方面涉及给药治疗有效量的这些化合物以治疗由神经功能障碍引起的病情、失调和疾病。
本发明的另一方面涉及用于本发明的组合物的给药方案,诸如从1天至约14天或从1天至约30天的日剂量的给药,更优选地,从约7天至约24天,以及最优选地,从约12天至约16天的日剂量的给药。
本发明的另一优选的方面涉及组合物以及通过给药治疗有效量的l-4-氯犬尿氨酸来治疗抑郁和治疗包括痛觉过敏的各种类型的疼痛的方法。
在另一个方面,本发明涉及如本申请中描述的药物组合物以及相关的治疗方法,该治疗方法给药产生7-氯犬尿喹啉酸的血药水平的量的l-4-氯犬尿氨酸。本发明的优选的方面涉及足以产生在约15ng/ml至550ng/ml的范围内的7-氯犬尿喹啉酸的血药水平的剂量的l-4-氯犬尿氨酸的给药。
本发明的另一方面涉及组合药物产品和相关的方法,包括包含l-dopa和本申请中描述的l-4-氯犬尿氨酸的药物组合物的制剂,以及涉及它们的共同给药,以本申请中描述的剂量同时或依次给药,以减少与dopa相关联的运动障碍。
本文中引用的所有参考文献,包括专利申请和出版物,它们的全部内容结合于此作为参考。
附图说明
下面描述了以下的图,并且以下的图合并入说明书中且构成说明书的一部分,以下的图示出了根据本发明的示例性实施例并且不应认为限制本发明的范围,因为本发明可以承认其他等同的有效实施例。这些图不必按比例绘制,并且为了清楚和简明,可以按比例放大或示意地示出某些图和和图的某些视图。
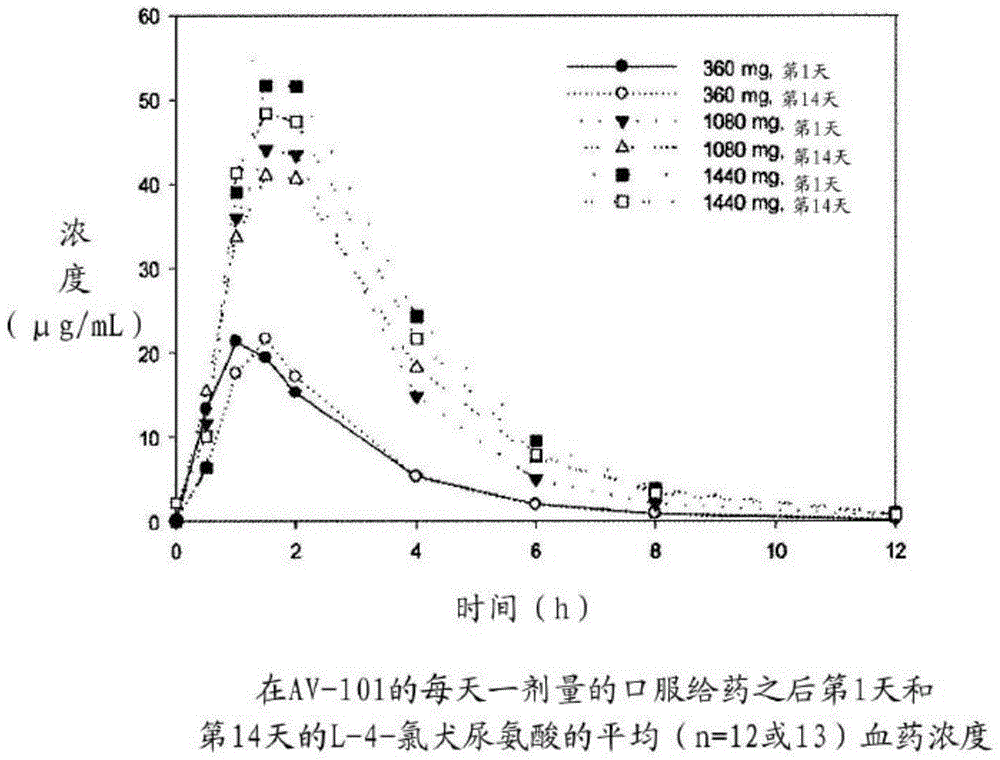
图1示出了在每日一次口服给药l-4-氯犬尿氨酸之后第1天和第14天的l-4-氯犬尿氨酸的平均(n=12或13)血药浓度。
图2示出了在每日一次口服给药l-4-氯犬尿氨酸之后第1天和第14天的7-氯犬尿喹啉酸的平均(n=12或13)血药浓度。
具体实施方式
下面参考详细的说明性实施例描述了本发明。显而易见地,本发明可以以各种各样的形式体现,其中一些形式可以与公开的实施例的那些大相径庭。因此,下面公开的特定结构和功能细节仅是代表性的,并且不限制本发明的范围。
在由国家卫生研究院的款项支持的工作中,已经示出,l-4-氯犬尿氨酸(也称为vistagenav-101)在外围组织炎症和神经损伤的三种动物模型中具有有效的抗痛觉过敏作用,而没有副作用的迹象。总的说来,在动物模型中,在动物模型中产生抗痛觉过敏的效果的剂量下,l-4-氯犬尿氨酸耐受良好,并且未产生安全毒性。
本发明基于发明人的临床发现:特定剂量的l-4-氯犬尿氨酸实际上在人体中是安全的,并且是可忍受的,而没有实质的副作用或任何显著的不利影响。此外,出人意料地发现,在这样的剂量下,患者报告减轻了神经性疼痛,尤其是痛觉过敏疼痛。该化合物是抗痛觉过敏的。也出人意料地发现,临床研究中的相当多的患者报告给药l-4-氯犬尿氨酸后的“幸福”的积极感受,“幸福”的积极感受是抗抑郁活性的一个指标,而这未被安慰剂对照报告。
本发明涉及组合物和治疗诸如由以下导致的各种类型的神经障碍、神经病(中枢和外周的)和功能障碍的治疗方法:(a)诸如由化疗和抗病毒药物引起的损伤和药物毒性;(b)诸如糖尿病、癌症、病毒感染、多发性硬化症、脊柱炎、多发性神经炎、手术、截肢、癫痫、抽搐、帕金森病、亨廷顿病、阿尔茨海默病以及涉及通过n-甲基-d-天门冬氨酸受体的过度活跃的谷氨酸能突触传递的病情的疾病和神经退行性疾病;和(c)与抑郁和其他精神障碍关联的神经递质、受体和信号路径的失衡。疼痛的治疗是明确地预期的,包括但不限于神经性疼痛、自发性疼痛、触诱发痛觉过敏疼痛、机械痛觉过敏疼痛和热痛觉过敏疼痛。此外,作为本发明的一个方面,也明确地预期抑郁的治疗。
定义:
基本上由特定量的药物活性试剂组成是指没有额外的量的试剂。不排除其他组分(例如,赋形剂和/或润滑剂)的存在或组合的不同药物活性的组分的存在。例如,明确地预期l-dopa和4-氯犬尿氨酸的组合,同其他组合物和用于它们的给药的方法一样,包括4-氯犬尿氨酸和同时或随后在一定时间范围提供的另一活性组分,其中,如同它们以单个组合药用产品给药,它们具有基本上相同的治疗效果。
“药物单位剂量”、“单位剂量”或“单位剂型”是指单剂量的l-4-氯犬尿氨酸,单剂量的l-4-氯犬尿氨酸能够给药至患者,并且可以容易地处理和包装,仍然作为物理和化学稳定的单位剂量。
“治疗有效”是指给药并且转化为7-氯犬尿喹啉酸以用于下调nmda-r介导的信号传递的l-4-氯犬尿氨酸的量,该量在神经功能中足以产生临床改善,诸如降低神经性疼痛或增加幸福的感受。
“没有显著的不利影响”是指对于给药l-4-氯犬尿氨酸的基本上所有患者来说,他们将具有不超过由美国食品和药物管理局(fda)定义的“轻微”不良事件。fda将“轻微不良事件”定义为患者容易忍受、导致最低程度的不适和不干扰日常活动的事件。相反,“中等不利事件”是不舒服至足以干扰正常的日常活动的事件。
药物组合物:
已经通过美国专利第5,547,991号,palfreyman等人的方法合成了l-4-氯犬尿氨酸。在医学文献中也已经报道了更新的合成工艺,诸如salituro等人在“enzyme-activatedantagonistsofthestrychnine-insensitiveglycine/nmdareceptor,”j.med.chem.1994;37-334,336中报道的。也可以从各种来源商业购买到l-4-氯犬尿氨酸,包括bocsciences(us,any,雪莉)和先进技术和工业有限公司(中国,香港)。在本专利申请中讨论的临床研究中使用了剑桥大学重点实验室(usa,wi,日耳曼敦)制造的l-4-氯犬尿氨酸。
本发明的优选实施例涉及药物组合物,该药物组合物包括用于与药学上可接受的载体和赋形剂一起口服给药而配制的l-4-氯犬尿氨酸的治疗有效量的单位剂量。
可以以包含根据本发明的l-4-氯犬尿氨酸并且产生如本申请中描述的7-氯犬尿喹啉酸的血药水平的任何药物形式配制本发明的药物组合物。应该想到,本发明描述的范围内的将给药的l-4-氯犬尿氨酸的精确剂量是安全和有效的,并且如本专利申请的图2和其他地方中描述的,它们产生由l-4-氯犬尿氨酸的给药产生的7-氯犬尿喹啉酸的血药水平。因此,明确地预期从约15ng/ml至约65ng/ml、从约65ng/ml至约300ng/ml以及从约300ng/ml至约550ng/ml的7-氯犬尿喹啉酸的血药范围。也应该想到,可以每天一次或多次给药本发明的单位剂量制剂,以延长4-氯犬尿氨酸水平升高至7-氯犬尿喹啉酸的治疗有效量的时间周期。
用于口服给药的l-4-氯犬尿氨酸的单位剂量药物组合物优选地包含约50mg至约1800mg,更优选地,约260mg至约1540mg,更优选地,约260mg至约460mg,约310mg至约410mg,约980mg至约1180mg,约1030mg至约1130mg,约1340mg至约1540mg,约1390mg至约1490mg,以及最优选地,约360mg、1080mg或1440mg。
应该想到,本发明的组合物的给药方案是治疗有效的。而如上所述,预期每天的给药方案,这将优选地为从约5天至约30天,包括由患者的医生确定的更短和更长的给药方案。特别地,明确地预期约7天至约24天和约12天至约16天的给药方案。
本发明的优选的方面涉及4-氯犬尿氨酸与l-dopa的联合给药以减小与l-dopa的维持剂量(通常地,对于每个患者,由医生凭经验确定)关联的运动障碍;或者以减小l-dopa的最小有效剂量,从而延迟发病和/或减小运动障碍的严重程度。根据本发明的药物组合物可以与l-dopa组合给药,同时或在时间接近上足够近以减轻l-dopa给药的副作用。用于l-dopa的给药的方案与运动障碍相关联是公知的。见,例如,tambasco,n.等人在“clinicalaspectsandmanagementoflevodopa-inducesdyskinesia,”parkinson’sdisease2012,articleid745947,doi:10.1155/2012/745947中报道的。
例如,根据本发明的药物组合物可以是片剂、胶囊、液体悬浮液、固体溶液、软胶囊、注射剂、局部或经皮或栓剂和鼻腔给药制剂。此外,本发明的药物组合物也可以是改进的释放形式,诸如但不限于双模形式或缓释形式。
通常地,可以通过药物制剂领域已知的传统的方法来制备本发明的药物组合物。例如,见remington的pharmaceuticalsciences第18版(mackpublishingcompany,easton,pa.,1990),其全部内容结合于此作为参考。在固体剂型中,l-4-氯犬尿氨酸可以与以下物质混合:至少一种药学上可接受的赋形剂,诸如柠檬酸钠或磷酸二钙,或(a)填充剂或增量剂,诸如淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸,(b)粘结剂,诸如纤维素衍生物、淀粉、藻酸盐、明胶、聚乙烯吡咯烷酮、蔗糖和阿拉伯树胶,(c)湿润剂,诸如甘油,(d)崩解剂,诸如琼脂、碳酸钙、马铃薯或木薯淀粉、藻酸、交联羧甲基纤维素钠、复合硅酸盐和碳酸钠,(e)溶液缓凝剂,诸如石蜡,(f)吸收促进剂,诸如季胺类化合物,(g)润湿剂,诸如鲸蜡醇和单硬脂酸甘油酯、硬脂酸镁等,(h)吸附剂,诸如高岭土和膨润土,以及(i)润滑剂,诸如滑石粉、硬脂酸钙、硬脂酸镁、固体聚乙二醇、月桂醇硫酸钠,或者它们的混合物。在胶囊、片剂和丸剂的情况下,剂型也可以包括缓冲剂。
在药物制剂领域中已知的药学上可接受的佐剂也可以用于本发明的药物组合物。这些佐剂包括但不限于保存剂、润湿剂、悬浮剂、甜味剂、香精、香料、乳化剂和分散剂。防止微生物的作用可以通过包括各种抗菌剂和抗真菌剂来确保,例如,对羟基苯甲酸酯、氯丁醇、苯酚、山梨酸等。也可以期望包括等渗剂,例如,糖、氯化钠等。如果需要,本发明的药物组合物也可以包含少量的辅助物质,诸如润湿剂或乳化剂、ph缓冲剂、抗氧化剂等,诸如柠檬酸、山梨糖醇酐单月桂酸酯、油酸三乙醇胺、丁基羟基甲苯等。
如上所述的固体剂型可以制备为具有包衣和壳体,诸如肠溶包衣和其他本领域公知的。它们可以包含安慰剂,并且也可以具有这样的组合物:以延迟的方式在肠道的特定部分释放一种或多种活性化合物。可以使用的嵌入组合物的非限制实例是聚合物质和蜡。活性化合物也可以是微胶囊的形式,如果合适的话,具有一种或多种上述赋形剂。
除了活性化合物之外的悬浮物可以包含悬浮剂,诸如异硬脂醇聚氧乙烯醚、聚氧乙烯山梨醇、山梨醇酯、微晶纤维素、偏氢氧化招、膨润土、琼脂、黄芪胶或这些物质的混合物等。
根据本发明,例如,用于直肠给药的组合物是可以通过混合结晶染料木素钠二水合物制备的栓剂,该栓剂具有例如合适的非刺激性赋形剂或载体,诸如可可脂、聚乙二醇或栓剂蜡,它们在常温下可以是固体,但是在体温下可以是液体,并且因此,在合适的体腔中溶解并且将活性组分释放在体腔中。
临床研究:
涉及健康的男性和女性受试者中的av-101的多种口服剂量进行1b阶段、单位点、随机、双盲、安慰剂对照的研究。受试者随机分成三组(360mg、1080mg和1440mg)并且连续14天接受每天的口服剂量。每组最初包括活性药的12个受试者和安慰剂的4个受试者。评估av-101对辣椒素诱导的痛觉过敏的安全性、药代动力学(pk)、治疗的耐受性和抗痛觉过敏作用。
以下pk参数来源于血药浓度与时间曲线以确定av-101(l-4-氯犬尿氨酸)和活性代谢产物7-氯犬尿喹啉酸的单剂量和多剂量pk曲线:最高浓度(cmax)、除半衰期(t1/2)、最高浓度的时间(tmax)、从时间0到最后可测量浓度的时间的血浆研究药物浓度与时间曲线下的面积(auc0-t)和从时间0外推至无穷大的血浆研究药物浓度与时间曲线下的面积(auc0-∞)。
将血液收集在6ml肝素锂真空采血管中。离心样本,并且从细胞分离血浆且在离心之后30分钟内冷冻。在将离心的样本放置在冷冻箱之前,将它们放置在冰上。在将血浆样本运输至分析人员之前,在约-20℃下冷冻保存血浆样本。
在第1天和第14天给药之前收集基线样本(0分钟)。然后在第1天和第14天服药之后的0.5小时、1小时、1.5小时、2小时、4小时、6小时、8小时、12小时和24小时处收集样本。
具有串联质谱分析的液相色谱用于确定人体血浆中的7-氯犬尿喹啉酸和l-4-氯犬尿氨酸。7-氯犬尿喹啉酸的标准曲线范围为从2.00ng/ml至1000ng/ml,定量的下限为2.00ng/ml。l-4-cl-kyn的标准曲线范围为从0.05μg/ml至50μg/ml,其中定量的下限为0.05μg/ml。分析均使用50.0μl的血浆样本量。
在研究中完全以剂量的范围来表征av-101的pk。在单次或多次给药之后获得l-4-氯犬尿氨酸和7-氯犬尿喹啉酸的血药浓度-时间曲线,360mg、1080mg或1440mg的每天一次口服剂量与口服剂量的快速吸收和分析物的一阶消除一致,证明了多室动力学,尤其是对于代谢产物7-氯犬尿喹啉酸。l-4-氯犬尿氨酸的平均tmax值随着剂量水平的增大而增大,对于最高剂量组达到接近2个小时。平均t1/2值在剂量之间相当一致,在从1.64小时到1.82小时的范围内。虽然不成比例,l-4-氯犬尿氨酸的平均cmax和auc0-∞值呈现为几乎是剂量线性的。第1天的平均cmax值在从360mg剂量之后的27.7μg/ml至1440mg剂量之后的64.4μg/ml的范围内。第1天的平均auc0-t值在从最低剂量之后的64μg·h/ml至1440mg剂量之后的196μg·h/ml的范围内。第14天时的大多数时间的平均cmax和auc0-t值稍微低于第1天的值。
通常地,如预期那样,代谢产物7-氯犬尿喹啉酸的最高浓度与l-4-氯犬尿氨酸的最高浓度同时出现或迟于l-4-氯犬尿氨酸的最高浓度出现,7-氯犬尿喹啉酸的平均tmax值在从1.67小时至2.34小时的范围内。这在图1和图2中示出。平均t1/2值在从2.52小时至3.23小时的范围内,平均t1/2值比l-4-氯犬尿氨酸的平均t1/2值在剂量之间变化稍微更大。平均t1/2值不呈现为是剂量相关的。7-氯犬尿喹啉酸的平均cmax和auc0-t值也呈现为几乎是剂量线性的。第1天的平均cmax值在从360mg剂量之后的42.7ng/ml至1440mg剂量之后的314ng/ml的范围内。第1天的平均auc0-t值在从最低剂量之后的156ng·h/ml至1440mg剂量之后的985ng·h/ml的范围内。类似于母体化合物,第14天的7-氯犬尿喹啉酸的平均cmax和auc0-t值通常稍微低于第1天的值。
实例1:l-4-氯犬尿氨酸对辣椒素诱导的痛觉过敏的抗痛觉过敏作用。
在临床研究的第1天和第14天,将250μg辣椒素的两次皮内注射依次注射入交替前臂的掌侧以产生灼烧疼痛、二级痛觉过敏和耀斑。辣椒素usp(美国药典)根据网站的标准步骤制备并且以10mg/ml的浓度溶解在20%的环糊精中。
在av-101或安慰剂的口服给药之后1小时在一个前臂中进行第一次辣椒素注射,并且在av-101或安慰剂的给药之后2小时在另一个前臂中进行第二次辣椒素注射。感觉神经测试在每次辣椒素注射时立即开始。在注射前以及在每次辣椒素注射之后0分钟、5分钟、10分钟、15分钟、30分钟、45分钟和60分钟处使用100mm直观模拟量表(vas)进行系列疼痛评估。检查人要求受试者通过使用通过施加5.18机械刺激、40℃探针以及用1英寸的泡沫刷温柔轻抚引起的疼痛和自发性疼痛的vas评价强度。vas由在0mm末端处写有“无疼痛”和在100mm末端处写有“最严重的疼痛”的100mm线构成。以毫米计的距离提供疼痛测量。
通过从不产生疼痛的皮肤区域以逐渐接近的半径朝着疼痛区域的中心正切地移动,直到受试者报告疼痛或压痛,由此来确定针对5.18机械刺激的痛觉过敏区域的边界。使用相同的评估并且以不同的角度开始来作出痛觉过敏区域边界的至少八个决定。此外,要求受试者评价在研究药物给药之后4.5小时(±5分钟)处对大腿前侧施加的1分钟45℃热刺激(短暂的热刺激)的疼痛强度,然后是每30分钟(±5分钟)至在研究药物给药之后6小时。施加辐射温度探针以在疼痛评估期间将皮肤温度固定在36℃。对于所有疼痛评估,受试者均使用vas。
所有研究评估时间点均锚定至时间0、研究药物给药的时间。由于在服药之后1小时处进行辣椒素注射,评估间隔(即,在服用研究药物之后的时间)为60分钟至120分钟,在此期间,在约60分钟、65分钟、70分钟和75分钟处进行评估,然后是每15分钟至在120分钟的时间点(即,在辣椒素注射之后0分钟、5分钟、10分钟、15分钟、30分钟、45分钟和60分钟)。在服用临床试验材料(ctm)之后约2小时进行辣椒素的第二次注射,并且评估间隔(即,在服用ctm之后的时间)为120分钟至180分钟。如在第一次辣椒素注射之后所述的,系列疼痛评估遵循相同的时间表。
主要疗效终点为在第14天服药之后120分钟至180分钟的av-101的每个剂量水平处对自发性疼痛的镇痛反应。对于治疗组和安慰剂组之间的自发性疼痛评估,疼痛时间曲线(aupc)下面的面积没有显著改变。同样地,对于任何的次要疗效终点,治疗组和安慰剂组之间没有显著的改变(对于在第1天服药之后120分钟至180分钟的时间间隔内的自发性疼痛的aupc;和对于自发性疼痛、来自机械刺激的引发疼痛与在第1天和第14天服药之后60分钟至180分钟的时间间隔内的来自40℃探针的引发疼痛的aupc)。然而,本发明发现,对于接受1080mgav-101的受试者(组2)和接受安慰剂的受试者之间的触诱疼痛、机械痛觉过敏疼痛和热痛觉过敏疼痛来说,aupc的最小二乘均数一致减小。在表1中示出了这些数据。
表1:疼痛评估分数
实例2:l-4-氯犬尿氨酸的抗抑郁活性
本申请发明人也出人意料地发现l-4-氯犬尿氨酸的增强情绪或抗抑郁活性。在本申请中描述的临床研究中,26个受试者中的5个(与安慰剂组中的0个受试者形成对比)肯定地报告幸福的感受。这与谷氨酸能系统有助于抑郁的病理生理学以及压力可以引起nmda受体的变化的报道一致。例如,见calabrese等人在“stress-inducedchangesofhippocampalnmdareceptors:modulationbyduloxetinetreatment,”plosone2012,7(5):e37916.doi:10.1371/journal.pone.0037916中的报道。
总之,如上所述,本申请中描述的发明总的来说涉及l-4-氯犬尿氨酸的剂型以及用于治疗神经疾病的方法,其中该神经疾病以通过n-甲基-d-天门冬氨酸受体介导的过度活跃的谷氨酸能突触传递表征。虽然以上详细地描述了和在附图中示出了特定的示例性实施例,但是应该理解,这样的实施例仅在于说明而不在于限制广义的发明。特别地,应该认识到,本发明的教导应用于各种各样的疾病。本领域普通技术人员将认识到,在不背离广义的发明范围的情况下,可以对以上描述的本发明的示出的实施例和其他实施例作出各种更改。因此,将理解,本发明不限于公开的特定实施例或布置,而旨在覆盖如附加的权利要求限定的本发明的范围和精神内的任何变化、改编或更改。
参考文献:
本申请中提及的以下期刊论文和所有其他出版物、专利和文本的全部内容结合于本发明作为参考。
(1)carteraj.glycineantagonist:regulationofthenmdareceptorchannelcomplexbythestrychnine-insensitiveglycinesite.drugsfuture1992;17:595-613.
(2)catarzietal.,competitivegly/nmdareceptorantagonists,curr.top.med.chem.2006;6(8):809-21.
(3)hokarim,wuh-q,schwarczr,smithqr.facilitatedbrainuptakeof4-chlorokynurenineandconversionto7-chlorokynurenicacid.neuroreport1996;8(1):15-18.
(4)kempja,fosterac,leesonpd,priestleyt,tridgettr,iversenll,etal.7-chlorokynurenicacidisaselectiveantagonistattheglycinemodulatorysiteofthen-methyl-d-aspartatereceptorcomplex.procnatlacadssciu.s.a.1988;85(17):6547-6550.
(5)lees-c,schwarczr.excitotoxicinjurystimulatespro-drug-induced7-chlorokynurenateformationintheratstriatuminvivo.neurosciencelett2001;304(3):185-188.
(6)leesonpd,iversenll.theglycinesiteonthenmdareceptor:structure-activityrelationshipsandtherapeuticpotential.jmedchem1994;37(24):4053-4067.
(7)linderholm,etal.,activationofratventraltegmentalareadopamineneuronsbyendogenouskynurenicacid:apharmacologicalanalysis,neuropharmacology2007;53(8):918-924.
(8)parsonscg,danyszw,quackg,hartmanns,lorenzb,wollenburgc,etal.novelsystemicallyactiveantagonistsoftheglycinesiteofthen-methyl-d-aspartatereceptor:electrophysiological,biochemicalandbehavioralcharacterization.jpharmacolexpther1997;283(3):1264-1275.
(9)raots,graynm,dappenms,clerja,micksj,emmettmr,etal.indole-2-carboxylates,novelantagonistsofthen-methyl-d-aspartate(nmda)-associatedglycinerecognitionsites:invivocharacterization.neuropharmacol1993;32(2):139-147.
(10)rundfeldtc,wlazp,loscherw.anticonvulsantactivityofantagonistsandpartialagonistsforthenmdareceptor-associatedglycinesiteinthekindlingmodelofepilepsy.brainres1994;653(1-2):125-130.
(11)wuhq,leesc,scharfmanhe,schwarczr.l-4-chlorokynurenineattenuateskainate-inducedseizuresandlesionsintherat.expneurol2002;177(1):222-232.
(12)wuh-q,lees-c,schwarczr.systemicadministrationof4-chlorokynureninepreventsquinolinateneurotoxicityintherathippocampus.eurjpharm2000;390:267-274.
- 还没有人留言评论。精彩留言会获得点赞!