一种计算缺陷团簇基态构型的方法
本发明属于核材料辐照损伤模拟技术领域,具体涉及一种计算缺陷团簇基态构型的方法。
背景技术:
辐照损伤一般指的是高能粒子对材料的辐照产生一系列缺陷的过程,辐照损伤严格来说,是由于种子、带电粒子或电磁波等和固体材料的点阵原子发生一系列碰撞,引起材料内部出现大量原子尺度的缺陷的过程。这些缺陷经过长时间的迁移、聚集和复合等会形成缺陷团簇、空洞等。
总的来说,无论是在高能粒子轰击材料诱导的级联碰撞中,还是在后续微结构演化的缺陷聚集过程中,都会形成空位和自间隙原子的缺陷团簇(vn/sian)。而获得vn/sian的基态结构对于进一步计算团簇的能量学和动力性性质以及预测微结构演化都至关重要。目前计算vn/sian基态结构时,一种是基于穷举法,即在一定解域内穷举所有可能的构型;一种是通过动力学方法探索体系势能面,或者基于生物群智能算法(如差分进化,de),全局探索体系势能面。
目前的计算方法所能研究的团簇尺寸n基本上越来越大,但是由于在计算不同备选构型的能量时都需要对原子尺度模型体系进行弛豫,进而计算总能,较为耗时间。当团簇尺寸n超过一定程度时(如100),往往即使采用群智能优化方法也变得不太可行。
技术实现要素:
有鉴于此,本发明有必要提供一种计算缺陷团簇基态构型的方法,该方法基于离散化模型对缺陷团簇基态构型进行计算,可高效地获得材料中较大型空位与自间隙团簇的基态构型,节约计算时间的同时,拓展了团簇尺寸上限。
为了实现上述目的,本发明采用以下技术方案:
本发明提供了一种计算缺陷团簇基态构型的方法,其包括以下步骤:
s1、设置采用de方法优化团簇构型时的缺陷团簇尺寸n0、以及结合离散化模型时优化的团簇构型尺寸上限n1;
s2、优化缺陷团簇构型,判断优化的缺陷团簇尺寸n是否大于n0,若是,则弛豫缺陷团簇构型;若否,则重复步骤s2;
s3、建立缺陷团簇能量与局域结构的离散关系,获得缺陷团簇能量的离散化模型;
s4、根据所述离散化模型计算个体能量,优化缺陷团簇构型;
s5、判断优化后的缺陷团簇尺寸n是否大于n1,若是,则弛豫缺陷团簇基态构型,获得缺陷团簇尺寸在1-n1之间缺陷团簇的构型和能量;若否,则返回步骤s3迭代。
进一步的,步骤s1中,所述缺陷团簇尺寸采用弛豫法与离散模型法优化获得,其中,n0设置为30~50,n1设置为50~200。
进一步的,步骤s2中,所述优化缺陷团簇构型采用de方法,所述de方法基于弛豫方法计算缺陷团簇构型。
进一步的,所述弛豫方法为共轭梯度法。
进一步的,步骤s3具体包括以下步骤:
计算缺陷团簇配位环境和缺陷团簇自身近邻关系,其中,所述缺陷团簇配位环境包括第一近邻原子数na1、第二近邻原子数na2、第三近邻原子数na3,所述缺陷团簇自身近邻关系包括第一近邻缺陷数nv1、第二近邻缺陷数nv2、第三近邻缺陷数nv3;
线性拟合缺陷团簇形成能
根据拟合结果精度,增删表征结构的离散量个数,最终建立缺陷团簇能量的离散化模型。
进一步的,所述第一近邻原子数na1为满足式|rvi-raj|<disn1的原子数,其中,rvi为构成缺陷团簇的对象i的坐标,raj为缺陷团簇周围原子j坐标,disn1为第一近邻距离;
计算所述第二近邻原子数na2、第三近邻原子数na3时,将disn1分别换成对应的第二近邻距离disn2、第三近邻距离disn3即可,其中,对于体心立方金属,
所述第一近邻缺陷数nv1为满足式|rvi-rvj|<disn1的缺陷数,其中,rvi、rvj分别为构成缺陷团簇的对象i、j的坐标;
计算所述第二近邻缺陷数nv2、第三近邻缺陷数nv3时,将disn1换为第二近邻距离disn2、第三近邻距离disn3即可。
进一步的,步骤s4中,所述的根据所述离散化模型计算个体能量,具体为:对于其中的每个个体计算其所在配位环境{na1,na2,na3,nv1,nv2,nv3},基于公式
与现有技术相比,本发明具有以下有益效果:
本发明通过建立小型缺陷团簇能量与其局域结构的离散化关系,从而可高效地获得材料中较大型空位与自间隙团簇的基态构型。具体的说,同现有的模拟方法相比,在群智能优化团簇构型中避免了直接弛豫计算团簇能量,节约了计算时间,拓展了基于弛豫计算体系能量的优化方法所能优化的团簇尺寸的上限。
附图说明
图1为本发明中一较佳实施方式中计算缺陷团簇基态构型方法的主要步骤流程框图;
图2为本发明中图1中计算缺陷团簇基态构型方法的算法流程图;
图3为本发明应用实施例1中采用de方法计算得到的v1-v24空位团簇构型;
图4为本发明应用实施例1中采用de方法计算得到的v25-v40空位团簇构型;
图5为本发明应用实施例1中采用de方法计算得到的v1-v40空位团簇的形成能
图6为本发明应用实施例1中结合de方法和空位团簇离散化能量模型计算得到的v41-v56空位团簇构型;
图7为本发明应用实施例1中结合de方法和空位团簇离散化能量模型计算得到的v57-v72空位团簇构型;
图8为本发明应用实施例1中结合de方法和空位团簇离散化能量模型计算得到的v73-v88空位团簇构型;
图9为本发明应用实施例1中结合de方法和空位团簇离散化能量模型计算得到的v89-v104空位团簇构型;
图10为本发明应用实施例1中结合de方法和空位团簇离散化能量模型计算得到的v105-v120空位团簇构型;
图11为本发明应用实施例1中结合de方法和空位团簇离散化能量模型计算得到的v41-v120空位团簇的形成能
具体实施方式
为了便于理解本发明,下面将结合具体的实施例对本发明进行更全面的描述。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明的公开内容理解的更加透彻全面。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本发明。
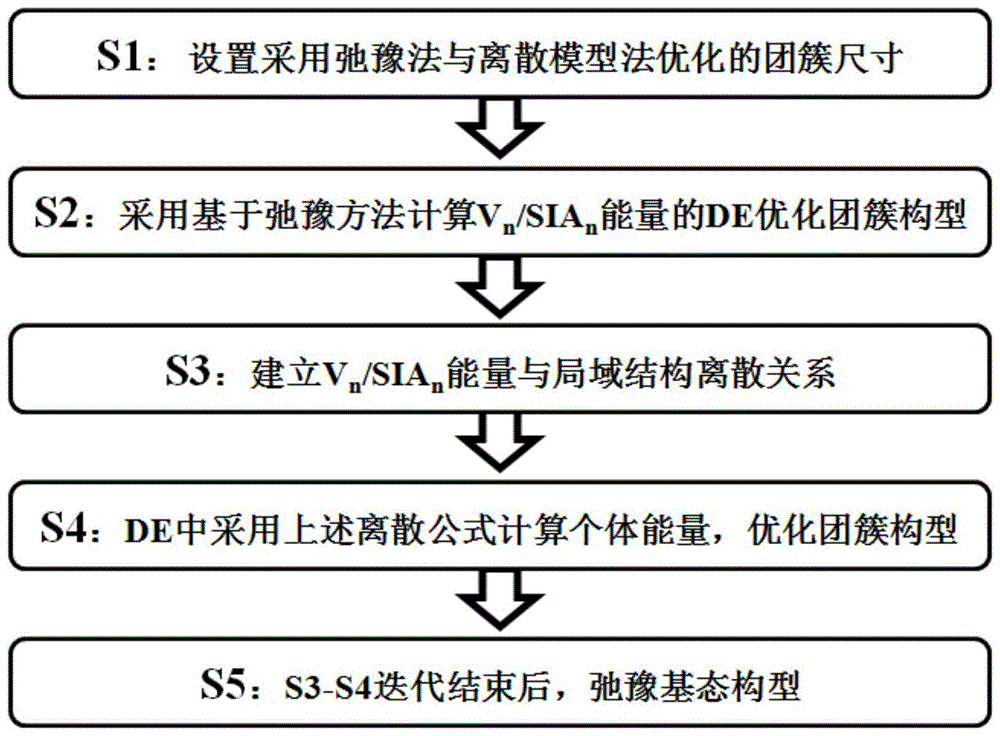
本发明中公开了一种计算缺陷团簇基态构型的方法,其具体步骤如图1中所示的,包括以下步骤:
s1、设置缺陷团簇尺寸n0、n1,其中,n0为采用de方法优化团簇构型时的缺陷团簇尺寸,n1为结合离散化模型时优化的团簇构型尺寸上限;
s2、优化缺陷团簇构型,判断优化的缺陷团簇尺寸n是否大于n0,若是,则弛豫缺陷团簇构型;若否,则重复步骤s2;
s3、建立缺陷团簇能量与局域结构的离散关系,获得缺陷团簇能量的离散化模型;
s4、根据所述离散化模型计算个体能量,优化缺陷团簇构型;
s5、判断优化后的缺陷团簇尺寸n是否大于n1,若是,则弛豫缺陷团簇基态构型,获得缺陷团簇尺寸在1-n1之间缺陷团簇的构型和能量;若否,则返回步骤s3迭代。
进一步的,请继续参阅图1,步骤s1中,所述缺陷团簇尺寸采用弛豫法与离散模型法优化获得,由于弛豫法与离散模型法获得缺陷团簇尺寸为现有的常规方法,因此,这里不再具体阐述。其中,由于n0过小会导致拟合数据太小,外推效果不佳;而n0太大,则会使得de计算较多,因此,为了控制计算量同时保证拟合效果达到一定精度,本发明优选的,将n0设置为30~50。而鉴于de方法的局限性,本发明的一些优选的实施方式中,将n1设置为50~200。
进一步的,步骤s2中,优化团簇构型可以采用本领域中任意的常规方式,优选的,在本发明中一些具体的实施例中,所述优化缺陷团簇构型采用de方法,所述de方法基于弛豫方法计算缺陷团簇构型。
优选的,所述弛豫方法为共轭梯度法,可以理解的是,弛豫方法并不局限于本发明中采用的共轭梯度法,本领域中其他常规的弛豫方法也可以适用于本发明。
进一步的,步骤s3具体包括以下步骤:
计算缺陷团簇配位环境和缺陷团簇自身近邻关系,其中,所述缺陷团簇配位环境包括第一近邻原子数na1、第二近邻原子数na2、第三近邻原子数na3,所述缺陷团簇自身近邻关系包括第一近邻缺陷数nv1、第二近邻缺陷数nv2、第三近邻缺陷数nv3;
线性拟合缺陷团簇形成能
根据拟合结果精度,增删表征结构的离散量个数,最终建立缺陷团簇能量的离散化模型,需要说明的是,近邻原子量和近邻缺陷量的选择原则为:分别从na1-na3和nv1-nv3中随机选择一定数量的拟合变量,选取使得模型拟合精度大于0.9的最少变量组合。
进一步的,所述第一近邻原子数na1为满足式|rvi-raj|<disn1的原子数,其中,rvi为构成缺陷团簇的对象i的坐标,raj为缺陷团簇周围原子j坐标,disn1为第一近邻距离;
计算所述第二近邻原子数na2、第三近邻原子数na3时,将disn1分别换成对应的第二近邻距离disn2、第三近邻距离disn3即可,其中,对于体心立方金属,
所述第一近邻缺陷数nv1为满足式|rvi-rvj|<disn1的缺陷数,其中,rvi、rvj分别为构成缺陷团簇的对象i、j的坐标;
计算所述第二近邻缺陷数nv2、第三近邻缺陷数nv3时,将disn1分别换为对应的第二近邻距离disn2、第三近邻距离disn3即可。
进一步的,步骤s4中,所述的根据所述离散化模型计算个体能量,具体为:对于其中的每个个体计算其所在配位环境{na1,na2,na3,nv1,nv2,nv3},基于公式
请继续参阅图2,图2示出了图1中一较佳实施方式中所述的计算方法的算法流程图,其具体过程如下:
s101、设置缺陷团簇尺寸n0和n1,其中,n0设置在30-50,n1设置在50-200,缺陷团簇尺寸采用弛豫法与离散模型法优化获得;
s102、调用基于弛豫方法计算缺陷团簇构型的de方法优化缺陷团簇构型;
s103、判断缺陷团簇尺寸n是否大于n0,若是,则进入步骤s104;若否,则返回步骤s102;
s104、n>n0,弛豫缺陷团簇构型;
s105、分析缺陷团簇能量与局域结构的离散关系,建立离散化模型;
s106、调用de方法并基于离散化模型计算个体能量,优化缺陷团簇构型;
s107、判断缺陷团簇尺寸n是否大于n1,若是,则进入步骤s108;若否,则返回步骤s105;
s108、n>n1,弛豫缺陷团簇构型,获得缺陷团簇尺寸在1-n1之间缺陷团簇的构型和能量。
下面结合具体的应用实施例对本发明中的计算缺陷团簇基态构型的方法进行更加清楚完整的说明。
应用实施例
本实施例中的金属材料为铁,其晶格常数a为
(1)设置参数n0、n1分别为40、120;
(2)采用de计算1-40空位团簇结构和能量,如图3-5所示,并建立团簇能量与配位环境关系,其中,图3-图4为采用de方法计算得到的v1-v40空位团簇构型,图3和图4中小球代表一个空位,直线连接处于第二近邻的两个空位。图5为采用de方法计算得到的v1-v40空位团簇的形成能
(3)采用de并结合步骤s2中能量的离散化模型,计算41-120团簇构型及其能量,如图6-11所示,其中,图6-图10为结合de方法和空位团簇离散化能量模型计算得到的v41-v120空位团簇构型,图11为结合de方法和空位团簇离散化能量模型计算得到的v41-v120空位团簇的形成能
通过应用实施例的示例可以看出,通过本发明中计算缺陷团簇基态构型的方法,建立小型缺陷团簇能量与其局域结构的离散化关系,从而可高效地获得材料中较大型空位与自间隙团簇的基态构型,节约了计算时间,拓展了基于弛豫计算体系能量的优化方法所能优化的团簇尺寸的上限。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!