大规模单细胞转录组数据高效聚类方法
本发明属于数据挖掘技术领域,具体涉及一种单细胞转录组聚类方法,可用于大规模细胞类型识别,细胞轨迹推理,基因差异表达。
背景技术:
随着单细胞测序技术的发展和测序成本的下降,越来越多的大规模单细胞数据被产生。而对这些大规模高维的单细胞数据进行处理和分析是一个非常具有挑战性的工作。聚类分析是单细胞分析中最基础的一个步骤,聚类的准确率和效率对后续分析有很大的影响。
以往的单细胞聚类算法,由于当时数据量的限制,在设计算法时,并没有考虑到应用于大规模的数据,其主要是应用于小规模的单细胞数据集。当数据量提升时,其运行时间会大幅增加,效率是十分低的,甚至有的算法不能正常运行。
目前已经开发了一些专门的聚类算法来从单细胞rna数据集中识别细胞的类型。
2015年chenxu等人在bioinformatics上提出的单细胞聚类方法snn-cliq,是利用共享最近邻snn的概念定义相似性并进行构图,之后使用一种基于准clique的聚类算法进行聚类。这个方法在高维度的单细胞基因表达数据集上,比传统方法更有优势,此外,它需要很少的输入参数,且能够自动确定聚类的个数。
2016年justinazurauskiene等人在bmc上提出了一个名为pcareduce的聚类算法,它整合了主成分分析pca和层次聚类方法,建立了主成分分析给出的降维后的表示和细胞聚类的数量之间的关系。
2017年peijielin等人在genomebiology上提出了一个名为cidr的聚类算法,该算法通过插补以减少单细胞数据中dropout的影响,并且改进了pca和层次聚类算法,在当时聚类精度方面优于当时最先进的算法,t-sne,zifa和raceid,cidr通常在几秒钟内可以完成处理数百个细胞的数据集,几分钟内完成处理数千个细胞的数据集,这在当时可以说是最快的算法。
2017年bowang等人在naturemethods上提出的名为simlr的分析框架和软件,它主要的贡献是从单细胞rna-seq数据中学习出一个合适的度量细胞间相似性的标准,有了这种相似性度量标准后,就可以进行后续的降维、聚类和可视化。
2017年vladimiryu.kiselev等人在naturemethods上提出了一个单细胞rna测序数据的无监督聚类算法sc3。它是利用多个度量标准计算细胞之间的距离,构建距离矩阵后通过k-means聚类,再将多个距离度量标准得到的聚类结果通过cspa进行一致聚类得到最终的聚类结果。通过一致聚类的方法将多个聚类结果组合到一起,从而得到高精度和鲁棒的聚类结果。但这个算法需要研究人员自己定义聚类个数,但在大多数情况下,研究人员并不能确切的知道需要聚几类。
2018年debajyotisinha等人在nucleicacidsresearch上提出的名为dropclust的聚类算法。这个算法是利用局域敏感哈希技术,开发的一种适用于大规模单细胞rna数据的聚类算法,其在运行时间和效率上显著优于其他聚类算法。
2018年andrewbutler等人在naturebiotechnology发表的seurat工具,是现在单细胞分析中使用最广泛的工具之一,其中的聚类分析使用的是snn-cliq图聚类的改进版。
以上算法都是偏向于对小于40000个小规模细胞进行聚类,但是在现实中,细胞数量是远远大于40000,而这些算法在大于40000个大规模细胞的数据中均存在运行时间慢,效率低,甚至不能正常工作,即计算机不能正常运行的不足。
技术实现要素:
本发明的目的在于克服上述现有技术的不足,提出一种大规模单细胞转录组数据高效聚类方法,以提高在大规模数据中运行时间和效率,并保证在对大规模数据进行聚类时,计算机能正常运行。
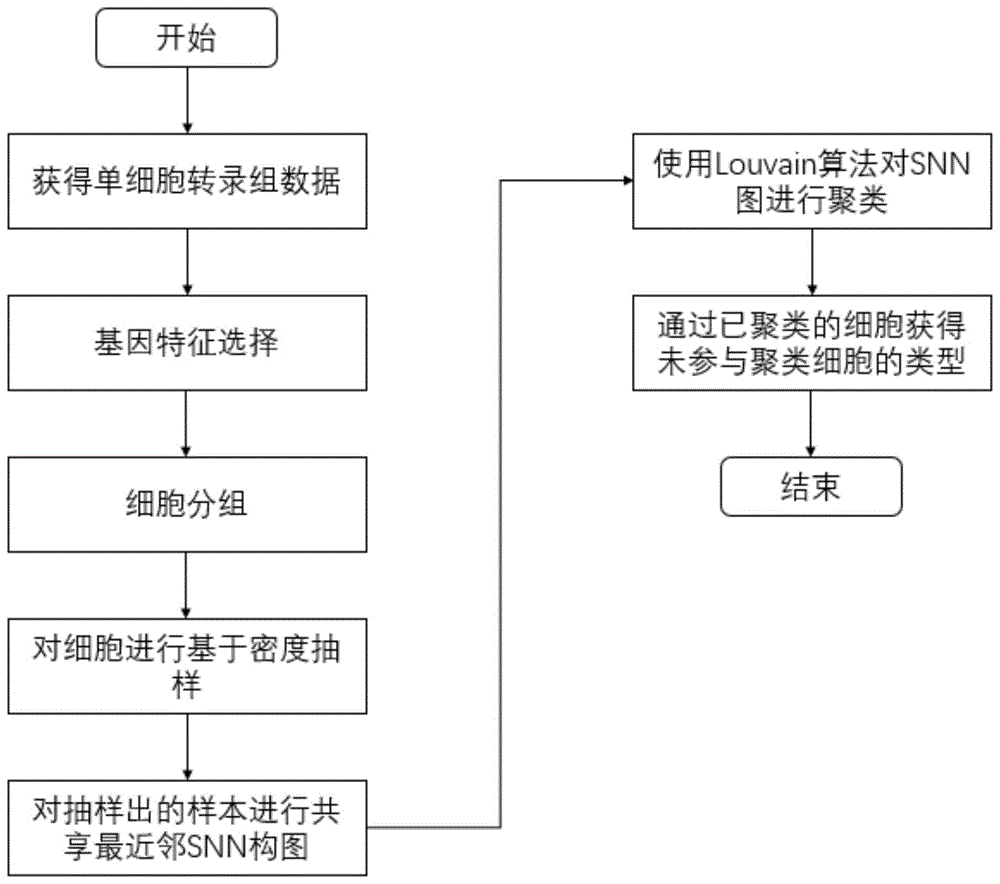
本发明的技术方案是:首先对单细胞转录组数据中的基因进行特征筛选,之后对单细胞转录组数据进行随机分组和密度抽样,接着对抽样出的新样本进行共享最近邻snn构图,然后使用louvain算法对图进行聚类,最后将聚类后的结果传播到未参与聚类的细胞。
其实现步骤包括如下:
(1)使用单细胞转录组测序技术对样本中的每一个细胞测量其基因表达值,产生单细胞转录组数据;
(2)对单细胞转录组数据中的基因进行特征选择,即从数据的基因中筛选出方差最大的前1000个基因作为高表达基因;
(3)按如下规则对单细胞转录组数据中的细胞进行随机分组:
当细胞数小于2000时,将所有细胞分为1组;
当细胞数大于2000而小于20000时,将2000个细胞分成一组,并在所有组内进行抽样,总共抽取2000个细胞;
当细胞数大于20000时,将2000个细胞分成一组,并在所有组内进行抽样,总共抽取1/10细胞数量的细胞;
(4)对分组后的细胞进行密度抽样:
(4a)对于每一组样本通过近似最近邻搜索算法hnsw查找近似k近邻,得到组内每个细胞的近似k近邻,并计算组内每个细胞到其近似k近邻距离之和sumd(ci),ci为原始样本中的细胞;
(4b)以组内每个细胞的近似k近邻距离和sumd(ci)作为抽样的概率,对每一组细胞进行概率抽样;
(4c)将每组样本中抽样出的细胞合并为一个新样本;
(5)按如下规则对抽出的新样本进行共享最近邻snn构图:
(5a)通过近似最近邻查找,找出新样本中每个细胞的近似k近邻;
(5b)对新样本中的每个细胞c′i′查找与其拥有相同近邻的细胞c′j′,并在该细胞c′i′与细胞c′j′之间连接一条边e(c′i′,c′j′),构成共享最近邻snn图;
(6)使用louvain算法对共享最近邻snn图进行聚类,得到抽样出细胞的细胞类型;
(7)对于剩下的未参与聚类的细胞,分别在已知标签的细胞中查找近似k近邻,k近邻中最多的那一类细胞的类型,即为未参与聚类的细胞类型。
本发明与现有技术相比,具有如下优点:
1)本发明由于在聚类的时候进行了密度抽样,不用对所有数据进行处理,只对抽样出的细胞进行聚类,因而相比现有直接对所有细胞进行聚类的方法,不仅减少了运行的时间,而且对日后产生的更大的数据集也能高效的运行;
2)本发明由于使用了密度抽样,因而相比于随机抽样,选出来的细胞更加全面,即使某一类型的细胞数量很少,也有更大的概率被选中;
3)本发明由于在进行共享最近邻snn图构建时使用近似最近邻搜索算法hnsw查找近似k近邻,因而相比传统的搜索k近邻的算法拥有更短的时间,更高的效率。
4)本发明由于在获取全部细胞类型时,是通过将已知的细胞类型传播到未参与聚类的细胞类型中去,因而相比直接对全部细胞进行聚类的方法更高效。
附图说明
图1为本发明的实现流程图。
具体实施方式
以下结合附图和具体实施例,对本发明作进一步详细说明。
本实施例以10000个细胞的单细胞转录组模拟数据为例。数据规模为10000细胞,30000个基因。
参照图1、本实例大规模单细胞转录组数据高效聚类方法,实现步骤如下:
步骤1,对单细胞转录组模拟数据的30000个基因进行特征筛选。
1.1)使用splatter软件包,生成10000个细胞,30000个基因的单细胞转录组模拟数据;
1.2)对单细胞转录组模拟数据中的30000个基因,通过如下公式计算每个基因的方差si,
其中,
1.3)将方差最大的1000个基因作为筛选出的高表达基因。
步骤2,对单细胞转录组模拟数据的细胞进行随机分组。
将10000个细胞随机分为5组,即每2000个细胞为一组;
步骤3,对分组后的细胞进行密度抽样。
3.1)对每一组的2000个细胞,通过近似最近邻搜索算法hnsw查找近似10近邻,得到每个组内细胞的近似10近邻细胞,并通过下式计算组内每个细胞到其近似10近邻距离之和sumd(ci):
其中ci为组内的细胞,
3.2)以每个细胞的近似10近邻之和sumd(ci)作为抽样的概率,对每一组细胞进行抽样,每组抽样出400个细胞,总共抽样出2000个细胞,作为新样本。
步骤4,对抽样出的2000个新样本进行共享近邻snn构图。
对抽样出的2000个细胞通过近似最近邻查找,找出每个细胞的10近邻,并对新样本中的每个细胞c′i′查找与其拥有相同近邻的细胞c′j′,并在细胞c′i′与细胞c′j′之间连接一条边e(c′i′,c′j′),构成共享最近邻snn图,该边通过下式计算:
其中,
步骤5,使用louvain算法对共享最近邻snn图进行聚类,得到新样本中2000个细胞的类型。
5.1)将共享最近邻snn图中的每个细胞设为一类,此时图中有2000类;
5.2)通过下式计算共享最近邻snn图的模块度q:
其中,m为图中边的总数量,wi′表示所有指向新样本中第i个细胞的连边权重之和,δ(c′i′,c′j′)为判别函数,并通过下式计算:
5.3)令第i个细胞不再属于自己的类型,变为与其细胞类型不同的细胞j的类型,计算细胞类型改变后的共享最近邻图snn新的模块度q′,
其中,c″i′是改变细胞类型后的第i个细胞,δ(c″i′,c′j′)为新判别函数,并通过下式计算:
5.4)通过下式计算模块度增量δq:
δq=q′-q;
5.5)将细胞i的细胞类型划分到细胞类型改变后共享最近邻图snn模块度增量δq最大,且最大的δq大于0的那个细胞类型中去,如果最大的δq小于0,则细胞类型不改变;
5.6)将同一类型的细胞聚合为一个细胞,返回5.1)进行迭代,直到共享最近邻snn图中所有细胞的类型不再发生变化。
步骤6,获得未参与聚类的8000个细胞的类型。
对于剩下的8000个未参与聚类的细胞,分别在已知标签的2000个细胞中查找近似10近邻,10近邻中最多的那一类细胞,即为该细胞的细胞类型。
本实例中,通过聚类获得10000个细胞的细胞类型。
以下结合模拟实验,对本发明的技术效果进行描述。
一.仿真条件:
仿真实验的计算机硬件cpu为intelcore(tm)i7、计算机硬件内存为16g;
计算机软件:windows10系统上rstudio集成开发软件。
二.仿真内容:
仿真1:用本发明与现有的3种方法seurat,cidr,sc3在5种不同细胞数量的单细胞转录组模拟数据中进行运行时间快慢的对比,结果如表1:
表14种方法在5种不同细胞数量的单细胞转录组模拟数据的运行时间表
从表1可以看出,本发明的运行时间都是最快的,并且随着数据量的增加,优势越明显,在10000个细胞数量时,已经为seurat方法运行时间的一半,而sc3方法在5000,10000个细胞的时候,已经不能正常在计算机上运行了,cidr方法在10000个细胞时也不能正常运行。
仿真2:用本发明与现有的3种方法seurat,cidr,sc3在4种单细胞转录组真实数据中进行准确性高低的对比,结果如表2:
表24种方法在4种单细胞转录组真实数据集中的准确性表
从表2可以看出,本发明在运行的准确性方面是仅次于sc3的方法,但从表1可知sc3方法的运行时间远远高过本发明,并且sc3方法需要人工给出聚类个数作为参数。
- 还没有人留言评论。精彩留言会获得点赞!