一种肺腺癌生物标志物筛选、预后模型构建及生物学验证的新方法
1.本发明涉及一种模型构建及生物学验证的新方法,特别涉及一种肺腺癌生物标志物筛选、预后模型构建及生物学验证的新方法。
背景技术:
2.目前,肺癌是一种死亡率和发病率居全世界首位的恶性肿瘤。除此之外,肺癌恶性程度高,预后较差,5年生存率仅有8%。肺癌的治疗手段多种多样,传统的治疗手段包括药物治疗、手术治疗、放疗、化疗等。进入二十一世纪后,分子靶向治疗取得了重大突破,通过针对癌症特异性因子进行治疗的效果远远好于传统疗法。因此,找到与癌症的发病机理相关的特异性生物分子也是当前的肿瘤生物学领域的研究热点。
3.近几年越来越多的研究团队致力于肺腺癌肿瘤标志物的挖掘和预后模型的构建,这些方法可以分为两类,一类是基于传统生物学的方法:研究者们采集肺癌患者的尿液,血浆,病理切片,细胞组织液等生物样本,通过分析实验组和对照组中不同生物分子的浓度,或者通过抗原
‑
抗体结合反应、相关激酶代谢反应、免疫学反应或者与某种特定试剂的阳性反应来判断。这样生化方法通常手段复杂,实验花费开销昂贵,效率低,同时会由于实验人员的操作带来误差。另一类是基于高通量组学与统计学、机器学习结合的标志物挖掘方法。常规的方法包括以医学统计学为基础,最常见的是cox变量分析寻找与预测目标相关性较强的标志物,并使用km曲线进行生存分析挖掘标志物的方法。同时,生信数据库在标志物的筛选中也发挥重要作用,例如david,string,genebank,gene ontology等被广泛地应用于生物信息的挖掘中。随着机器学习和数据挖掘技术的发展,越来越多的方法被应用到标志物的筛选中。常规的rfe特征选择算法,二进制的粒子群优化算法,深度学习也在不同的数据集体现出色性能,同时使用机器学习算法取代传统的风险回归模型也能的都更高的精度。虽然基于高通量的手段可以减小实验损耗,提升效率,但是面对上万维度的特征时,如何有效地选出最佳标志物这一问题,依然是当前相关领域要解决的重点难题。
4.递归特征消除的主要思想是反复的构建模型,然后根据特征重要性筛选删除一部分不重要的特征,接下来在剩余的特征上重复这个过程,直到当前特征集合为空为止。之后输出被删除的特征排序即为即为特征重要性排序。在生物学信息中,svm与rfe结合是最常用的方法,在多个生物组学数据集中表现优越。
5.sffs算法,即序列浮动向前算法是一种常见的基于wrapper的特征选择方法,基本思想如下:从空集开始,在未选择的特征中选择一个特征x,使子集加入x后评价函数达到最优。然后在已选择的特征中删除一个特征y,使子集剔除 z后评价函数达到最优。
技术实现要素:
6.本发明的目的是能快速、准确地筛选与肺腺癌患者预后生存相关的标志物,利用选出的标志物构建预后模型并对这些标志物进行生物信息学验证,而提供的一种肺腺癌生
物标志物筛选、预后模型构建及生物学验证的新方法。
7.本发明提供的肺腺癌生物标志物筛选、预后模型构建及生物学验证的新方法,其方法包括如下步骤:
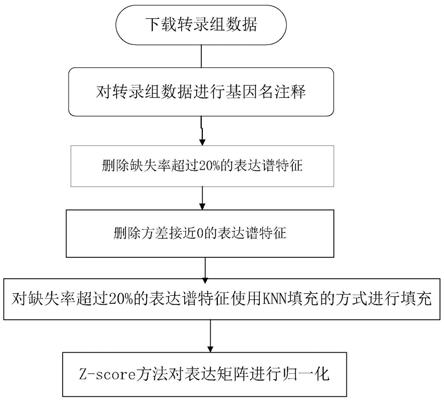
8.步骤一、对原始的基因表达矩阵进行数据的预处理,首先使用匹配文件进行注释,将探针名改为基因名,按照如下方式进行数据预处理:
9.1)、删除缺失率超过20%的转录组特征基因;
10.2)、删除方差接近0的转录组特征基因;
11.3)、对缺失率超过20%的转录组特征基因使用knn填充的方式进行填充;
12.4)、使用z
‑
score方法对上述3)得到的表达矩阵进行归一化;
13.最终,得到了一个含有数个基因,数个样本的转录组表达数据矩阵;
14.步骤二、在经过预处理后得到的数据上进行生物标志物筛选,流程如下所示:
15.1)、使用双边t检验去除癌症/非癌症组p值大于0.05的特征基因,p值越大,说明在不同分组种显著性差异越不明显;
16.2)、获取差异表达基因,即利用fold
‑
change检验计算出癌症/非癌症组的 logfc值和fdr值,保留|logfc|>1.5,fdr<0.05的基因,并且根据|logfc|的正负来判断该基因在癌症发生过程中是上调还是下调;
17.3)、利用scikt
‑
learn中的selectfrommodel模块来实现基于模型的特征选择,利用模型自带的特征评估的功能,删除低于某一特征评分的设定值的特征,该过程是一个迭代的过程,通过阈值和迭代次数的设定确定最终的结果;
18.4)、构建基因表达的相关性网络,并与fold
‑
change和generank算法相结合,调整svmrfe中的特征权重来确定最终的基因排序列表,改进的svmrfe 算法过程如下所示:
19.首先依据互信息公式构造一个互信息矩阵,互信息公式(1)如下所示:
[0020][0021]
将互信息矩阵转成拓扑重叠网络的形式,这一步的目的是寻找基因间的广泛联系:
[0022][0023]
按照公式(3)、(4)计算评分矩阵,将评分矩阵看成相关性网络:
[0024][0025][0026]
对相关性网络使用generank算法,按照公式(5)计算每个节点的重要性,这里相关性网络上的节点表示矩阵中的基因:
[0027][0028]
根据公式(5)的结果和支持向量机递归特征清除重新确定转录组中基因特征的重
要权重;
[0029]
采用svmrfe算法通过模型训练样本,对每个特征进行评分排序,去掉评分最低的特征,然后重复构建模型进行下一次训练,该过程中特征重要性的评估方法如公式(6):
[0030]
c
i
=w
i2
ꢀꢀꢀꢀꢀ
(6)
[0031]
支持向量机训练过程求出(6)之后,利用公式(4)重新计算每一个基因的重要性,此时的重要性为公式(5)和公式(6)两个结果的比值,如公式(7)所示:
[0032]
rank
i
=w
i2
/r
i[n]
ꢀꢀ
(7)
[0033]
经过公式(7)得到的结果为每一个基因重新计算的重要性。以此重要性为评估指标,进行递归特征清除算法;
[0034]
5)、输出执行改进的svmrfe算法得到的基因排序,选取前50个特征基因中的最优子集,在利用动态sffs方法对最优子集进行去冗余,得到的基因组合即可被认定为是最佳基因组合;
[0035]
去冗余的过程是一种改进的sffs算法,该算法首先从空集开始,从未被选中的特征中选择一个加入特征集合,使加入后分类准确率更高;然后从已选中的特征中删除特征,在该过程中使用一个数组记录当前特征数为i时的最佳分类准确率arr[i],如果删除过程中arr[i]升高,则继续删除,检验arr[i
‑
1] 的值,一直删除到该值不再继续升高为止;
[0036]
步骤三、使用经过上述多步骤特征选择的肺腺癌预后标志物构建预后模型,以生存期是否超过三年将其分成正、负样本,通过多种机器学习分类模型进行对比,使用五折交叉验证评估,用acc值,auc值,f1
‑
score值作为评估指标,选择线性支持向量机作为预后分类模型;
[0037]
支持向量机是去找到一个超平面,尽可能地将两个不同类别的样本分开,定义这个超平面为w
t
x+b=0,在二维平面中,就相当于直线w_1*x+w_1*y+b=0,其中,x代表输入向量,也就是样本集合中的向量;w是可调权值向量,每个向量可调权值;t代表向量的转置;b代表偏置,即超平面相对原点的偏移,而在超平面上方的点,定义为y=1,在超平面下方的点,定义为y=
‑
1,在支持向量机训练的过程中,实现任意一个样本与超平面的间隔最大化,这时的间隔称为硬间隔,该过程中的目标函数为:
[0038][0039]
由于的最大化等价于的最小化,因此根据凸优化理论,最终的目标函数为:
[0040]
在模型的构建中使用的是线性支持向量机,线性支持向量机,指原有的数据样本本可以寻找一个超平面使两个样本完全分离,但是混入了异常点导致无法线性可分或者由于异常点严重影响模型的泛化性能,在这种情况下,引入一个松弛变量ξ
i
,对应一个代价,使间隔函数加上松弛变量大于等于1,这个过程称为软间隔最大化,此时目标函数为:
[0041]
[0042]
在目标函数中,c表示惩罚系数,α
i
、μ
i
均为大于0的拉格朗日系数,c越大,对误分类的惩罚也越大;
[0043]
步骤四、建模之后对选出的标志物进行生信分析,其中包括基因组功能分析、km生存分析、通路分析和mirna靶基因分析,通路分析包括go分析、kegg 分析和reactome分析,生存分析是通过对患者的随访,分析一定时间后的患者生存或者死亡的状况,km生存分析中使用kaplan
‑
meier分析法。
[0044]
本发明的有益效果:
[0045]
本发明提供的肺腺癌生物标志物筛选、预后模型构建及生物学验证的新方法应用于tcga数据库的数据集中,能够筛选出45个基因。使用线性支持向量机模型验证45个基因标志物,利用五折较差验证的auc和acc作为模型评估指标,最终的结果为auc=0.98,acc=0.92。该结果明显优于大多数传统基因标志物选择方法。
[0046]
同时,本发明提出的方法中还包括了对标志物的生物学验证。从功能分析和靶基因的结果看出,选中的45个标志物有36个基因参与了与肺癌相关的基因调控过程,其他的标志物也被证实参与了癌症的发生和发展,将该标志物集合进行km分析和时间依赖的roc曲线,得到高低风险组p<0.00001的显著性差异和0.841的roc值。同时,本发明使用了包含通路分析,生存分析等方法进一步验证选中的标志物与肺癌相关的功能。在多种通路分析中,发现多种通路与肺癌的发生与发展有关。这也证明本发明不仅能找出与肺癌相关的标志物,准确预测肺癌患者的生存期,同时还能发掘与肺腺癌发生相关的通路,探究肺腺癌的发生机制。
附图说明
[0047]
图1为本发明所述的为肿瘤转录组数据的预处理流程示意图。
[0048]
图2为本发明所述的为肿瘤标志物的筛选流程示意图。
[0049]
图3为本发明所述的为预后模型的建立和生物信息的验证过程示意图。
具体实施方式
[0050]
请参阅图1至图3所示:
[0051]
本发明提供的肺腺癌生物标志物筛选、预后模型构建及生物学验证的新方法,其方法包括如下步骤:
[0052]
步骤一、对原始的基因表达矩阵进行数据的预处理,首先使用匹配文件进行注释,将探针名改为基因名。数据预处理的过程如下所示:
[0053]
1)、删除缺失率超过20%的转录组特征基因;
[0054]
2)、删除方差接近0的转录组特征基因;
[0055]
3)、对缺失率超过20%的转录组特征基因使用knn填充的方式进行填充;
[0056]
4)、使用z
‑
score方法对3)得到的表达矩阵进行归一化
[0057]
最终,得到了一个含有57000个基因,513个样本的转录组表达数据矩阵。
[0058]
步骤二、在经过预处理后得到的数据上进行生物标志物筛选,流程如下所示;
[0059]
1)、使用双边t检验去除癌症/非癌症组p值大于0.05的特征基因;
[0060]
2)、获取差异表达基因,即利用fold
‑
change检验计算出癌症/非癌症组的 logfc
值和fdr值,保留|logfc|>1.5,fdr<0.05的基因;
[0061]
3)、利用scikt
‑
learn中的selectfrommodel模块来实现基于模型的特征选择,利用模型自带的特征评估的功能,删除低于某一特征评分的设定值的特征。该过程是一个迭代的过程,通过阈值和迭代次数的设定确定最终的结果。
[0062]
4)、构建基因表达的相关性网络,并与fold
‑
change和generank算法相结合,调整svmrfe中的特征权重来确定最终的基因排序列表。改进的svmrfe 算法过程如下所示:
[0063]
首先依据互信息公式构造一个互信息矩阵,互信息公式(1)如下所示:
[0064][0065]
将互信息矩阵转成拓扑重叠网络的形式,这一步的目的是寻找基因间的广泛联系:
[0066][0067]
按照公式(3)、(4)计算评分矩阵,将评分矩阵看成相关性网络:
[0068][0069][0070]
对相关性网络使用generank算法,按照公式(5)计算每个节点的重要性,这里相关性网络上的节点表示矩阵中的基因:
[0071][0072]
根据公式(5)的结果和支持向量机递归特征清除重新确定转录组中基因特征的重要权重。
[0073]
svmrfe算法是一种基于支持向量机的包装式后向序列特征选择方法,该算法通过模型训练样本,对每个特征进行评分排序,去掉评分最低的特征,然后重复构建模型进行下一次训练。该过程中特征重要性的评估方法如公式(6)。
[0074]
c
i
=w
i2
ꢀꢀꢀꢀ
(6)
[0075]
支持向量机训练过程求出(6)之后,利用(4)重新计算每一个基因的重要性,此时的重要性为(5)和(6)两个结果的比值,如公式(7)所示。
[0076]
rank
i
=w
i2
/r
i[n]
ꢀꢀ
(7)
[0077]
经过公式(7)得到的结果为每一个基因重新计算的重要性。以此重要性为评估指标,进行递归特征清除算法。
[0078]
5)、输出执行改进的svmrfe算法得到的基因排序,选取前50个特征基因中的最优子集,在利用动态sffs方法对最优子集进行去冗余,得到的基因组合即可被认定为是最佳基因组合。
[0079]
去冗余的过程是一种改进的sffs算法,该算法首先从空集开始,从未被选中的特征中选择一个加入特征集合,使加入后分类准确率更高;然后从已选中的特征中删除特征,
在该过程中使用一个数组记录当前特征数为i时的最佳分类准确率arr[i]。如果删除过程中arr[i]升高,则继续删除,检验arr[i
‑
1] 的值,一直删除到该值不再继续升高为止。
[0080]
步骤三、使用经过上述多步骤特征选择的肺腺癌预后标志物构建预后模型,以生存期是否超过三年将其分成正、负样本,通过多种机器学习分类模型进行对比,使用五折交叉验证评估,用acc值,auc值,f1
‑
score值作为评估指标,选择线性支持向量机作为预后分类模型;
[0081]
支持向量机是去找到一个超平面,尽可能地将两个不同类别的样本分开,定义这个超平面为w
t
x+b=0,在二维平面中,就相当于直线w_1*x+w_1*y+b=0,其中,x代表输入向量,也就是样本集合中的向量;w是可调权值向量,每个向量可调权值;t代表向量的转置;b代表偏置,即超平面相对原点的偏移,而在超平面上方的点,定义为y=1,在超平面下方的点,定义为y=
‑
1,在支持向量机训练的过程中,实现任意一个样本与超平面的间隔最大化,这时的间隔称为硬间隔,该过程中的目标函数为:
[0082][0083]
由于的最大化等价于的最小化,因此根据凸优化理论,最终的目标函数为:
[0084]
在模型的构建中使用的是线性支持向量机,线性支持向量机,指原有的数据样本本可以寻找一个超平面使两个样本完全分离,但是混入了异常点导致无法线性可分或者由于异常点严重影响模型的泛化性能,在这种情况下,引入一个松弛变量ξ
i
,对应一个代价,使间隔函数加上松弛变量大于等于1,这个过程称为软间隔最大化,此时目标函数为:
[0085][0086]
在目标函数中,c表示惩罚系数,α
i
、μ
i
均为大于0的拉格朗日系数,c越大,对误分类的惩罚也越大;
[0087]
步骤四、建模之后对选出的标志物进行生信分析,其中包括基因组功能分析、km生存分析、通路分析和mirna靶基因分析,通路分析包括go分析、kegg 分析和reactome分析,生存分析是通过对患者的随访,分析一定时间后的患者生存或者死亡的状况,km生存分析中使用kaplan
‑
meier分析法。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1