一种抗感染药物注射剂的有效性评价方法及其应用与流程
1.本发明涉及药物评价技术领域,尤其涉及一种抗感染药物注射剂的有效性评价方法及其应用。
背景技术:
2.抗感染药物注射剂有效性研究的应用场景目前为仿制药质量与疗效一致性评价工作,已经上市药物评价/再评价工作中。对于注射剂而言,尤其是静脉注射剂大多认为直接进入血液,同品种相同规格的注射剂通常应具有相同的有效性水平。
3.目前,抗感染药物注射剂的有效性评价通常以药物制剂的体内药时曲线为基础进行评价,对于单个抗感染药物制剂的有效性评价,大多采用药时曲线与敏感菌的mic90或mic50进行比较评价;对于不同来源的同品种规格制剂间的比较,大多采用药动学参数例如c
max
、auc、t>mic等进行比较,以药时曲线对应药动学参数的80~125%作为一致性评判标准。
4.但是,抗感染药物通常对应多个治疗目的器官,采用静脉血总体药时曲线评价,不能表征药物在治疗目的器官的分布;而且,大多数抗感染药物属于广谱抗菌药物,一种抗感染药物对应不同致病菌或敏感菌具有不同的抗菌活性,现有的有效性评价方法难以准确地对不同来源制剂间的有效性差异,特别是治疗的等效性进行评价。
技术实现要素:
5.针对现有技术存在的问题,本发明提供一种抗感染药物注射剂的有效性评价方法及其应用,实现对抗感染药物注射剂提供更准确的有效性评价,此外,本发明所提供的方法也能进一步指导该类药物制剂制备过程中的质量控制。
6.本发明采用以下技术方案:
7.本发明提供一种抗感染药物注射剂的有效性评价方法,以所述抗感染药物注射剂在至少一个其适应症对应感染器官组织中的药物浓度时间曲线作为评价基础。
8.传统的抗感染药物注射剂的有效性评价方法中,以静脉血中的药物浓度时间曲线为评价基础,但此时只能从生物等效性的角度评价抗感染药物注射剂的有效性。本发明首次提出以抗感染药物注射剂在至少一个其适应症对应感染器官组织中的药物浓度时间曲线为评价基础,基于此选择合适的药效学指标进行有效性评价,将药物在体内的吸收
‑
分布
‑
代谢
‑
排泄过程向不同的组织器官进行推进,实现了生物等效性到治疗等效性的深入,更能准确地反映同品种规格抗感染药物制剂间的有效性差异。
9.本发明中适应症对应感染器官组织包括但不限于肺部、肝脏、脾脏、心脏、脑部、肾脏和皮肤等,具体考察个数依据具体的抗感染药物等实际情况进行确定。优选地,根据所评价抗感染药物的适应症,建立敏感菌
‑
感染器官组织的对应关系,针对不同器官组织对应的感染特征建立待评价药物
‑
敏感菌
‑
感染器官组织的药效评价关联,以得到更准确的评价结果。
10.进一步地,本发明以所述抗感染药物注射剂在静脉血中的药物浓度时间曲线为基础,根据所述抗感染药物注射剂在人体内的吸收
‑
分布
‑
代谢
‑
排泄规律,从而获得所述抗感染药物注射剂在其适应症对应感染器官组织中的药物浓度时间曲线。
11.优选地,通过公式(1)计算获得所述抗感染药物注射剂在其适应症对应感染器官组织中的药物浓度时间曲线;
[0012][0013]
其中:c
t
为适应症对应的某一感染器官组织中的药物浓度,v
t
为该器官组织的容积,q
art
为进入该器官组织的血流速率,q
ven
为离开该器官组织的血流速率,c
art
为进入该器官组织的血药浓度,r为药物的全血
‑
血浆比,为药物在该器官组织中的组织
‑
血浆分配系数,由公式(2)得到
[0014][0015]
其中:p为分配系数,在非脂肪组织器官中为药物的正辛醇
‑
水分配系数,在脂肪组织器官中为可离子化药物的中性形式在植物油和水之间的分布系数;f
nlp
、f
plp
、f
wp
分别为血浆中中性脂类、磷脂、水的容积分数;f
nlt
、f
plt
、f
wt
分别为组织中中性脂类、磷脂、水的容积分数。
[0016]
实测所述抗感染药物注射剂在其适应症对应感染器官组织中的药物浓度时间曲线是很复杂繁琐且耗时的,而且往往对应器官组织为多个,因此通过计算获得所述抗感染药物注射剂在其适应症对应感染器官组织中的药物浓度时间曲线非常有必要。本发明公式(1)的微积分方程很好地反映了药物在各组织器官中的分布,因此本发明以所述抗感染药物注射剂的血药时间曲线为基础,通过公式(1)进行计算,可以较为简单快捷地获得准确的药物在其适应症对应感染器官组织中的药物浓度时间曲线。其中,对于常规实验动物和人体的v
t
、q
art
和q
ven
这三个生理参数可通过文献直接获得。
[0017]
上述技术方案中,所述抗感染药物注射剂的血药时间曲线可实测获得或通过pbpk(生理药代动力学)模型计算获得,相关的获取方法为已知技术,本发明不再赘述。
[0018]
现有技术中,抗感染药物的药效学指标主要采用对应致病菌或敏感菌的mic(最小抑菌浓度)进行表征。最为准确的抗感染药物对应致病菌的mic测定方法为药敏试验法。常规抗感染药物的有效性评价采用的品种
‑
敏感菌mic结果可参考由美国临床和实验室标准协会(clinical and laboratory standards institute,clsi)定期发布的最新版本的“抗微生物药物敏感性实验的执行标准”(performance standards for antimicrobial susceptibility testing)。
[0019]
本发明研究发现,当所述抗感染药物注射剂为时间依赖型药物时,将适应症对应感染器官组织中的药物浓度水平(由所述药物浓度时间曲线反映)与敏感菌的mic进行比较得到相应器官组织的t
组织
>mic,以此为一级评价指标进行有效性评价更合理准确,有助于药效研究以及治疗方案评价等。
[0020]
当所述抗感染药物注射剂为浓度依赖型药物时,以其在适应症对应感染器官组织
中,药物浓度大于敏感菌最小抑菌浓度的曲线(该药物在其适应症对应感染器官组织中的药物浓度时间曲线)下面积与敏感菌最小抑菌浓度的比值为一级评价指标。
[0021]
在抗感染药物注射剂有效性的评价过程中,为反映同品种规格抗感染药物制剂间的有效性差异,通常会选取参比制剂(参比制剂的选取参照《化学仿制药参比制剂遴选与确定程序》、《化学仿制药参比制剂遴选申请材料要求》),将待评价药物的评价指标与参比制剂的该指标进行比较,若待评价药物的评价指标在参比制剂该指标的80~125%范围内,则判定待评价药物与参比制剂有效性一致。在本发明所提供的评价方法中,评价基础变为药物在适应症对应感染器官组织中的药物浓度时间曲线,而适应症对应感染器官组织往往为多个,即会同时出现多个比较结果,此时若以每个结果都在80~125%范围内进行判定,既不准确也不直观。
[0022]
本发明经过研究发现,将各适应症对应感染器官组织的比较结果以及各敏感菌条件下的比较结果综合起来,得到待评价抗感染药物注射剂的有效性水平,通过该有效性水平数值的高低,可以直观且较为准确地得出该药物的有效性评价结果。
[0023]
具体地,当所述抗感染药物注射剂为时间依赖型药物时,其有效性水平进一步以二级评价指标进行评价,所述二级评价指标为所述抗感染药物注射剂的一级评价指标与参比制剂的一级评价指标的比值。
[0024]
优选地,当考察的适应症对应感染器官组织为多个时,所述有效性水平以各适应症对应感染器官组织中所述二级评价指标的均值进行计算,
[0025]
当考察的敏感菌为多个时,所述有效性水平为各敏感菌分别计算获得的有效性水平的乘积。
[0026]
在本发明的具体实施方式中,所述有效性水平可由公式(3)计算,
[0027][0028]
其中,e为有效性水平,n为考察的适应症对应感染器官组织的数量,t
供试制剂
‑
器官
>mic
敏感菌1
为供试制剂在某一器官组织中药物浓度大于敏感菌1最小抑菌浓度的维持时间,t
参比制剂
‑
器官
>mic
敏感菌1
为参比制剂在某一器官组织中药物浓度大于敏感菌1最小抑菌浓度的维持时间,t
供试制剂
‑
器官
>mic
敏感菌2
为供试制剂在某一器官组织中药物浓度大于敏感菌2最小抑菌浓度的维持时间,t
参比制剂
‑
器官
>mic
敏感菌2
为参比制剂在某一器官组织中药物浓度大于敏感菌2最小抑菌浓度的维持时间,以此类推。其中,将t
供试制剂
‑
器官
>mic
敏感菌1
与t
参比制剂
‑
器官
>mic
敏感菌1
相除时,对应的是同一考察的器官组织,以此类推。
[0029]
当所述抗感染药物注射剂为浓度依赖型药物时,其有效性水平进一步以二级评价指标进行评价,所述二级评价指标为所述抗感染药物注射剂的一级评价指标与参比制剂的一级评价指标的比值;
[0030]
优选地,当考察的适应症对应感染器官组织为多个时,所述有效性水平以各适应症对应感染器官组织中所述二级评价指标的均值进行计算,
[0031]
当考察的敏感菌为多个时,所述有效性水平为各敏感菌分别计算获得的有效性水平的乘积。
[0032]
在本发明的具体实施方式中,可由公式(4)计算,
[0033][0034]
其中,e为有效性水平,n为考察的适应症对应感染器官组织的数量,auc
供试制剂
‑
器官>mic敏感菌1
为供试制剂在某一适应症对应感染器官组织中药物浓度大于敏感菌1最小抑菌浓度的曲线下面积,auc
参比制剂
‑
器官>mic敏感菌1
为参比制剂在同一考察的器官组织中药物浓度大于敏感菌1最小抑菌浓度的曲线下面积,其余类推。
[0035]
进一步地,当所述有效性水平不低于80,则判定所述抗感染药物注射剂与参比制剂的有效性一致,反之则有效性不一致。
[0036]
在本发明一个具体实施方式中,所述抗感染药物注射剂为注射用头孢曲松钠,选取金黄色葡萄球菌作为敏感菌。注射用头孢曲松钠对金黄色葡萄球菌的mic为2~4mg/l,为了能客观全面地反映制剂间的有效性差异,选择对应感染器官组织中注射用头孢曲松钠浓度大于4mg/l的维持时间为评价指标。优选地,所述适应症对应感染器官组织包括肺部、肌肉、肝脏、脾脏、心脏、脑部、肾脏和皮肤。
[0037]
在本发明另一个具体实施方式中,所述抗感染药物注射剂为注射用头孢唑林钠,选取金黄色葡萄球菌和大肠埃希菌作为敏感菌。具体地,分别采用1mg/l和4mg/l作为对金黄色葡萄球菌和大肠埃希菌的最小抑菌浓度mic。优选地,所述适应症对应感染器官组织包括肺部、肝脏、脾脏、心脏、脑部、肾脏和皮肤。
[0038]
本发明还提供上述抗感染药物注射剂的有效性评价方法在注射用抗感染药物的制备及质量控制中的应用。通过上述评价方法准确获知了待评价药物与参比制剂之间的有效性差异,才能有效反馈到药物的研发和制备中,更好地督促获得质量可控可靠的抗感染药物注射剂。
[0039]
本发明提供了一种抗感染药物注射剂的有效性评价方法,以抗感染药物注射剂在至少一个其适应症对应感染器官组织中的药物浓度时间曲线为评价基础,基于此选择合适的药效学指标进行有效性评价,将药物在体内的吸收
‑
分布
‑
代谢
‑
排泄过程向不同的组织器官进行推进,实现了生物等效性到治疗等效性的深入,更能准确地反映同品种规格抗感染药物制剂间的有效性差异,对获得质量可控可靠的抗感染药物注射剂具有重要意义。
附图说明
[0040]
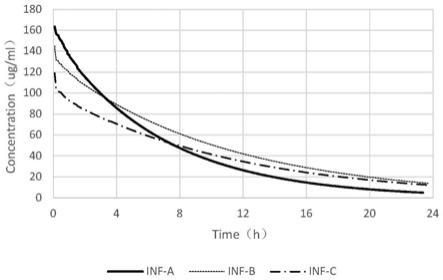
图1为本发明实施例1中三种不同试剂静脉血中的药时曲线;
[0041]
图2a~图2h为本发明实施例1中三种不同试剂在各适应症对应感染器官组织中的药物浓度时间曲线;
[0042]
图3为本发明实施例2中头孢唑林在人体静脉血中的药时曲线;
[0043]
图4为本发明实施例2中头孢唑林钠单次静脉给药1.0g不同组织器官的药物浓度时间曲线;
[0044]
图5a、图5b为本发明实施例2中头孢唑林钠单次静脉给药1.0g,每12小时给药一
次,共96小时的药物浓度时间曲线。
具体实施方式
[0045]
为使本发明实施例的目的、技术方案和优点更加清楚,下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0046]
实施例1
[0047]
本实施例提供一种注射用头孢曲松钠的有效性评价方法,具体如下:
[0048]
1.注射用头孢曲松钠的适应症、用法用量
[0049]
1.1适应症及对应组织器官:注射用头孢曲松钠主要用于治疗对本品敏感的致病菌引起的感染,如:脓毒血症;脑膜炎;播散性莱姆病(早、晚期);腹部感染(腹膜炎、胆道及胃肠道感染);骨、关节、软组织、皮肤及伤口感染;肾脏及泌尿道感染;呼吸道感染,尤其是肺炎、耳鼻喉感染;生殖系统感染等。
[0050]
1.2用法用量:成人通常采用静脉滴注,每24小时1~2g或每12小时0.5~1g。最高剂量一日4g,疗程7~14日。常规用量:1g/12hr。
[0051]
1.3药效学参数:对大肠埃希菌、肺炎克雷伯菌、产气肠杆菌、氟劳地枸橼酸杆菌、吲哚阳性变形杆菌、普鲁威登菌属和沙雷菌属的mic90介于0.12~0.25mg/l之间;对金黄色葡萄球菌的mic为2~4mg/l。
[0052]
2.头孢曲松的药代动力学参数与体内pk特征
[0053]
头孢曲松在人体内不被代谢,约40%的药物以原形自胆道和肠道排出,60%自尿中排出;肌内注射0.5g后24小时的血药浓度为6.0mg/l,血消除半衰期(t
1/2
)为7.1小时。1分钟内静注0.5g,即刻血药峰浓度(c
max
)为150.9mg/l,24小时后的血药浓度为9.9mg/l,血消除半衰期(t
1/2
)为7.87小时;头孢曲松主要分布到细胞外水分中,药物的分布体积呈现剂量依赖性;头孢曲松的血浆蛋白结合率呈现明显的浓度依赖性,正常人体结合率为95%;该药在中或弱肾功能患者体内的半衰期变化不大;与一些药物联合使用如丙磺舒等不能增高本品血药浓度或延长其半衰期。
[0054]
表1头孢曲松的药代动力学参数
[0055][0056]
3.头孢曲松pbpk模型搭建
[0057]
头孢曲松开展了大鼠、狗及人的pbpk模型搭建,计算不同剂型剂量给药后的体内pk曲线。头孢曲松同种属pbpk模型,单一参数组无法同时准确反映不同来源制剂的体内分布特征,表明该药物在体内分布受制剂影响较大。头孢曲松的eccs分类为class_3b,主要的清除机制为肾脏滤过清除;大鼠的肾脏清除采用0.05*kidney blood flow,肝脏清除为0.03l/h并全部定义为胆汁排泄;狗的清除采用0.1*kidney blood flow,肝脏清除为1.07l/h并全部定义为胆汁排泄;人的清除采用0.00555*kidney blood flow,肝脏清除为0.45l/h并全部定义为胆汁排泄;模型加载不同来源制剂对药物分布与清除的调整参数调整后,计算的pk曲线与观测值一致。
[0058]
4.获得注射用头孢曲松钠的药时曲线
[0059]
利用上述工作结果,获得注射用头孢曲松钠制剂单次静脉给药1.0g在人体内的药时曲线。此处取三个制剂分别命名为inf
‑
a、inf
‑
b和inf
‑
c(inf
‑
b为国家药监局于2019年8月27日发布的仿制药参比制剂目录第二十二批在列制剂,下称参比制剂;inf
‑
a与inf
‑
c均为国内企业生产的供试制剂,其中inf
‑
c制剂采用原研企业原料生产,inf
‑
a采用其他企业原料生产),给药剂量均为1.0g,给药途径均为静脉给药,血药浓度考察时间(t)为24小时。获得的药时曲线如图1所示。
[0060]
5.注射用头孢曲松钠不同制剂在各适应症对应感染器官组织中的药物浓度时间曲线及有效性评价结果
[0061]
利用公式(1),以三个制剂的药时曲线为基础,计算适应症对应各感染器官组织中的药物浓度时间曲线,其中适应症对应感染器官组织包括:肺部、肌肉、肝脏、脾脏、心脏、脑
部、肾脏和皮肤。
[0062]
根据头孢曲松对敏感菌的mic参数,由于本品种对大肠埃希菌、肺炎克雷伯菌、产气肠杆菌、氟劳地枸橼酸杆菌、吲哚阳性变形杆菌、普鲁威登菌属和沙雷菌属的mic90介于0.12~0.25mg/l之间,不同制剂在相应的组织器官中,差异较小不能客观全面反映制剂间的有效性差异,因此选择本品种对金黄色葡萄球菌的mic:2~4mg/l中的4mg/l为最小抑菌浓度,即以适应症对应感染器官组织中注射用头孢曲松钠浓度大于4mg/l的维持时间作为有效性评价指标,对三个制剂进行有效性评价。结果各适应症对应感染器官组织中的药物浓度时间曲线如图2a~图2h所示,各制剂在各器官组织中浓度大于4mg/l的维持时间如表2所示。
[0063]
表2三制剂在各器官组织中浓度大于4mg/l的维持时间
[0064][0065]
头孢曲松钠为时间依赖型抗感染药物,以公式(3)计算其有效性水平。
[0066]
结果inf
‑
a的有效性水平数值为33.3<80,inf
‑
c的有效性水平数值为89.2>80;根据该结果判定inf
‑
a与inf
‑
b的有效性不一致,inf
‑
c与inf
‑
b的有效性一致,即在治疗水平药效一致。
[0067]
结果比较
[0068]
若采用目前通用的制剂间有效性评价标准“对药代动力学参数在80.0%~
125.0%范围内认为相应制剂等效”,则三制剂的比较结果如表3所示。其中,注射剂有效性的体内药代动力学考察指标包括:c
max
和auc0‑
t
(即0至24小时的药时曲线下面积)。
[0069]
表3三制剂药时曲线对应药代动力学指标比较
[0070] inf
‑
ainf
‑
binf
‑
cc
max
(ug/ml)163.58144.874119.106%c
max
112.9% 82.2%auc0‑
t
(ug
·
h/ml)1080.51279.31043.9%auc0‑
t
84.5% 81.6%
[0071]
根据上表结果判断,inf
‑
a制剂与inf
‑
c制剂的相应药代参数均位于参比制剂inf
‑
b相应参数结果的80%~125%范围内,可判断为均与inf
‑
b制剂等效。
[0072]
从以上结果可以看出,以inf
‑
b为参比制剂,以传统评价方法,即在静脉血药时曲线水平,得出三个制剂的有效性一致,而以本发明的评价方法,可以得到inf
‑
c比inf
‑
a有效性更接近参比制剂的有效性,更有助于进一步的研发、生产或用药。
[0073]
实施例2
[0074]
本实施例提供一种注射用头孢唑林钠的有效性评价方法与给药方案评价,具体如下:
[0075]
1.注射用头孢唑林钠的适应症与敏感致病菌、给药方案(依据药品说明书)
[0076]
1.1适用于治疗敏感细菌所致的下列感染:
[0077]
呼吸道感染:肺炎链球菌、克雷伯菌属、流感嗜血杆菌、金黄色葡萄球菌(青霉素敏感和青霉素耐药)和a型β
‑
溶血性链球菌等。
[0078]
尿路感染:大肠埃希菌、奇异变形杆菌、克雷伯菌属、肠杆菌和肠球菌属细菌。
[0079]
皮肤及软组织感染:金黄色葡萄球菌(青霉素敏感和青霉素耐药)、a型b
‑
溶血性链球菌及其他链球菌属细菌。
[0080]
骨和关节感染:金黄色葡萄球菌。
[0081]
败血症:肺炎链球菌、金黄色葡萄球菌(青霉素敏感和青霉素耐药)、奇异变形杆菌、大肠埃希菌和克雷伯菌属。
[0082]
感染性心内膜炎:金黄色葡萄球菌(青霉素敏感和青霉素耐药)及a型b溶血性链球菌。
[0083]
肝胆系统感染:大肠埃希菌、各种链球菌属、奇异变形杆菌、克雷伯菌属及金黄色葡萄球菌。
[0084]
生殖系统感染:大肠埃希菌、奇异变形杆菌、克雷伯菌属及部分肠球菌。
[0085]
1.2成人用药:静脉推注、滴注或肌肉注射,一次0.5~1g,每日2~4次,严重感染可增加至一日6g,分2~4次静脉给予。
[0086]
1.3根据“抗微生物药物敏感性实验的执行标准”(m100
‑
s20版)中对标准菌种的敏感性数据,头孢唑林对金黄色葡萄球菌(atcc25923)的敏感度0.25~1ug/ml,对大肠埃希菌(atcc25922)的敏感度为1~4ug/ml。
[0087]
2.头孢唑林的药代动力学参数
[0088]
表4头孢唑林的药代动力学参数
[0089][0090]
3.头孢唑林pbpk模型的建立与药时曲线的获得
[0091]
头孢唑林主要经肾小球滤过及肾小管分泌,绝大部分(~80%)以原形排出,人体内的消除半衰期约为1.8小时;肾脏清除过程中,涉及摄取转运体oats和外排转运体mrp4的介导作用;除脑组织外,该物在全身可良好分布,在胆汁中的浓度较低,尿液中浓度较高;头孢唑林的血浆蛋白结合率呈现较明显的浓度依赖性,结合率可达74%~86%;药物在胃肠道吸收差,通常肌肉或静脉给药;在肾功能不全者体内半衰期延长,临床给药剂量应当调整。头孢曲松在人体内不被代谢,约40%的药物以原形自胆道和肠道排出,60%自尿中排出;肌内注射0.5g后24小时的血药浓度为6.0mg/l,血消除半衰期(t1/2β)为7.1小时。1分钟内静注0.5g,即刻血药峰浓度(cmax)为150.9mg/l,24小时后的血药浓度为9.9mg/l,血消除半衰期(t1/2b)为7.87小时。利用上述模型结合文献数据,分别进行头孢唑林在大鼠、狗及人等不同种属中pbpk模型的搭建,计算不同剂量、剂型给药后在体内的pk曲线。
[0092]
通过以不同种属药时曲线的验证与优化,注射用头孢唑林钠的pbpk模型中各参数及计算结果可以较准确地反映制剂在体内的吸收、分布、代谢、排泄过程。以注射用头孢唑林钠在人体内静脉给药1.0g的单剂量pbpk模型为基础,准确性参数分别为:r2=0.88、sse=728.6、rmse=9.543、mae=5.89。图3为头孢唑林在人体静脉血中的药时曲线,图中曲线为pbpk模型计算的药时曲线,方块为实测的血药浓度数据。
[0093]
4.头孢唑林钠各器官组织中的药效学评价
[0094]
图4为单次静脉给药1.0g不同器官组织的药物浓度时间曲线。分别对头孢唑林在肺部、肝脏、脾脏、心脏、脑部、肾脏和皮肤的药效水平进行考察,以单次给药12小时为药效考察的时间单元,分别通过t>mic的小时数和其与12小时的比值进行评价,结果见表5。根据不同组织器官适应症的敏感菌,将相应器官的药效学结果进行标记,结果显示,注射用头孢唑林钠在人体内单次静脉给药1.0g在12小时内的药效结果均不甚理想,以金黄色葡萄球菌和大肠埃希菌进行考察,药效水平均不及60%。
[0095]
表5金黄色葡萄球菌和大肠埃希菌在各组织器官中的t>mic和药效
[0096][0097]
根据药品说明书,对于非严重感染的治疗方案“静脉推注、滴注或肌肉注射,一次0.5~1g,每日2~4次”,采用1.0g的较高剂量以每日2次的频率进行连续给药,考察4日(96小时)内的药物浓度时间曲线,结果如图5a、图5b所示。
[0098]
通过连续给药的组织浓度考察,头孢唑林钠在12小时给药间隔中,无显著的药物浓度蓄积情况,药效水平与单次给药基本一致。提示在剂量为1.0g的静脉途径间隔12小时连续给药的治疗方案中,头孢唑林的治疗有效水平难以满足要求,建议对治疗方案进行修订与提高。
[0099]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1