基于高通量实验数据挖掘进行关键RNA功能挖掘的方法与流程
本发明涉及生物信息学,具体涉及基于高通量实验数据挖掘进行关键rna功能挖掘的方法。
背景技术:
人类基因组dna核苷酸序列中约93%能被转录为rna,其中仅2%的转录产物被翻译为蛋白质,余下98%属于非编码rna(ncrna)。随着microrna的研究进展,揭示了ncrna在人类基因转录后调节、细胞生长、分化、增殖中起着相当重要的作用。ncrna的研究热度最高的主要是microrna、circrna、lncrna。在肿瘤研究领域,mrna和ncrna的研究同样重要。近年来,生物信息方案层出不穷,在研究mrna和ncrna功能上,共表达关系和蛋白互作网络日益受到广泛应用。
临床肿瘤研究面临的一些难点:1)临床样品难以收集到足够的大规模数量,不利于统计和建模;2)有不少现有方法基于tcga等数据集,但是这些数据集存在不完全开放的问题,要下载原始数据需要非常多的权限,一般研究者们都没法申请到这些权限,以至于只能下载三级数据(处理并校正过的数据,非原始数据)等,不适合与tcga以外的临床数据进行联合分析;3)目前大型癌症lncrna表达谱分析发现各种肿瘤类型之间表现出转录水平的差异,显示出lncrna在疾病研究中巨大的挖掘潜力,lncrna可以被视做肿瘤组织中转录过程的“暗物质”,但是lncrna已知功能极少,欠缺比较全面的数据库帮助揭示功能机制,以至于经常遇到找到明显差异lncrna却不知道如何继续往下研究的问题。而经常遇到的问题还有,显著差异lncrna并不止一个,有很多个,研究者通常在这种情况下,希望先用生物信息的方法排除一些,并对重要性有排序,这样就可以在往下进行细胞功能研究的时候不至于茫无目标,像大海捞针一样。
如何从已有公开实验数据,特别是公开的高通量实验数据中挖掘出关键rna功能,是一项非常重要且有意义的工作。
技术实现要素:
本发明的目的在于克服现有技术的至少一个不足,提供一种基于高通量实验数据挖掘进行关键rna功能挖掘的方法。
本发明所采取的技术方案是:
基于高通量实验数据挖掘进行关键rna功能挖掘的方法,包括如下步骤:
s1)收集肿瘤高通量实验数据并进行背景处理及数据清洗,得到肿瘤rna表达数据;
s2)对肿瘤rna表达数据进行归一化处理;
s3)基于归一化处理后的数据,进行基因差异表达分析及数据集过滤;
s4)对过滤后的数据集进行相关性统计分析并选择p-value显著性阈值p<0.05的数据集,记为高相关数据集;
s5)对高相关数据集进行基因功能富集分析、rna生物通路分析、rna蛋白互作用网络分析,确定rna共表达功能网络,确定关键rna及其功能;
其中相关性统计分析具体包括:
建立rna相关性矩阵scor:
计算rna探针与本数据集中其他任意探针的pearson相关系数,从而得到相关性矩阵;
利用rankaggregation计算出rna探针与其他探针的相关性p-value,按照p-value数值从小到大排序,得到与rna探针所相关的其他探针的重要性排序,
按照相同的方法,计算k个实验里面的相关性排序,然后综合k个实验的各自结果得到每个探针相关性排序的总排名;
获得共表达关系权重评分sco:
根据p-value显著性阈值,提取显著差异rna的最相关的探针列表,并找出相应探针所对应的基因,即得到与rna相关的共表达基因。
在一些实例中,背景处理及数据清洗的操作包括:先使用工具对rna芯片数据进行背景信号,过滤掉芯片杂交信号中属于非特异性的背景噪音部分,完成背景处理后,对于负值和噪声信号,使用变异系数法和k邻近法进行数据清洗。
在一些实例中,使用r软件的affy/limmapackage工具对rna芯片数据进行背景信号。
在一些实例中,收集肿瘤高通量实验数据还包括将基因探针表达值转换为rna表达值,得到更全面的rna数据库,具体包括对基因探针进行基因组定位,并把rna序列映射到基因组上,通过位置叠加的关系,与整合的rna数据库进行交叠,找出基因探针对应的rna并将基因探针的表达值转化为rna表达值。
在一些实例中,转换rna表达值的原则如下:
1)如果一个探针只与一个rna转录本发生重叠,则rna转录本表达值=探针表达值;
2)如果一个探针与两个以上rna转录本发生重叠,且所处的正负链方向一致,则rna转录本1表达值(1v2)=探针表达值,rna转录本2表达值(1v2)=探针表达值;
3)如果一个rna转录本与两个以上探针发生重叠且基因组距离小于1000bp,则rna转录本表达值(2v1)=(探针1表达值+探针2表达值)*50%;
4)如果rna转录本表达值同时存在1v2和2v1的情况,则需要计算综合的rna转录本表达值=rna转录本表达值(2v1)-rna转录本表达值(1v2),作为最终的rna转录本表达值;
5)如果转换后的rna转录本表达值存在负值,则采用k邻近法进行数值校正。
在一些实例中,基因差异表达分析及数据集过滤具体包括:根据研究目的或实验设计,对多组样本两两之间进行对比,获取差异rna列表。
在一些实例中,临床检测样品数据量少于15对在选出初步的差异基因后,加入类似的伴随数据集进行对比,获取差异rna列表;所述伴随数据集为过滤得到的数据集,其过滤原则包括:1)与研究的临床特征相符;2)control/test的对照设计一致;3)根据临床数据集里rna表达量,找出初步差异rna,用这个初步差异rna的集合去计算候选已发表数据集中的相应rna的表达值标准差;如果标准差小于0.2,则认为初步筛选出来的差异rna在候选已发表数据集中不具备相似特性,不能入选伴随数据集,不能与临床检测样品一起进行分析。
在一些实例中,所述伴随数据集的来源选自1)ncbigeo;2)ncbisra;3)ena–ensemblenucleotidearchive中的至少一个。
在一些实例中,rna蛋白互作用网络分析使用的数据库基于多个开放蛋白互作数据库建立,即对多个数据库中的数据进行合并去冗余,并把多个数据库的综合评分相乘得到最终sppi。
在一些实例中,整合的数据库包括:igdb.nsclc数据库、scop数据库、dip数据库、string数据库、spike数据库、reactome数据库、pfam数据库、pdb数据库、mint数据库、intact数据库、hprd数据库、biogrid数据库。
在一些实例中,对高相关数据集还进行疾病-rna关系评分sdisease,包括确定rna在不同数据库出现的次数以及在同一个疾病描述中共同出现的rna;进一步的,使用的数据库包括:lncrnadisease、omim、ncbiclinvar数据库、hlungdb。
在一些实例中,对高相关数据集还进行调控因子tf关系评分stf:包括进行rna的cis或trans的targets预测及rna上游的tf预测,cis表示染色体邻近位置的靶点;trans表示不同染色体或染色体远端位置的靶点。
在一些实例中,根据连坐法形成最终的rna共表达功能网络。
在一些实例中,所述肿瘤为肺癌。
本发明的有益效果是:
本发明一些实例的方法,可以高效地从已有实验数据中成功挖掘关键rna功能,可从一大堆已知的rna中找出项目研究最相关的基因,同时可以对未知的rna进行功能的预测,从而更好的确定其在生命活动中所行使的角色,为后续疾病机制、药物靶点、疾病诊断等方面提供重要依据。
本发明一些实例的方法,对于标本收集难度大的肿瘤,例如小细胞肺癌,可以在公共数据库资源的基础上,借助本分析方法,增加样品量,使临床研究更有统计意义,同时让一些疾病临床基因模型建立成为可能,具有其创新性。
本发明一些实例的方法,专门整合了肺癌研究相关的多个数据库,包括肺癌基因位点数据库,肺癌基因-疾病关联数据库,肺癌基因-机理数据库这几类,对于研究肺癌各种信号途径的rna尤其有帮助。
本发明一些实例的方法,专门整合了多个肺癌基因研究相关的数据库,包括lugend、igdb.nsclc和hlungdb等等,方便肺癌研究者筛选肺癌中的高置信的致病、早筛、预后相关的rna,也可以根据自己的研究目的,对筛选出来的rna进行目的相关性的排序,帮助研究者更快找出临床样品的关键关联rna。
附图说明
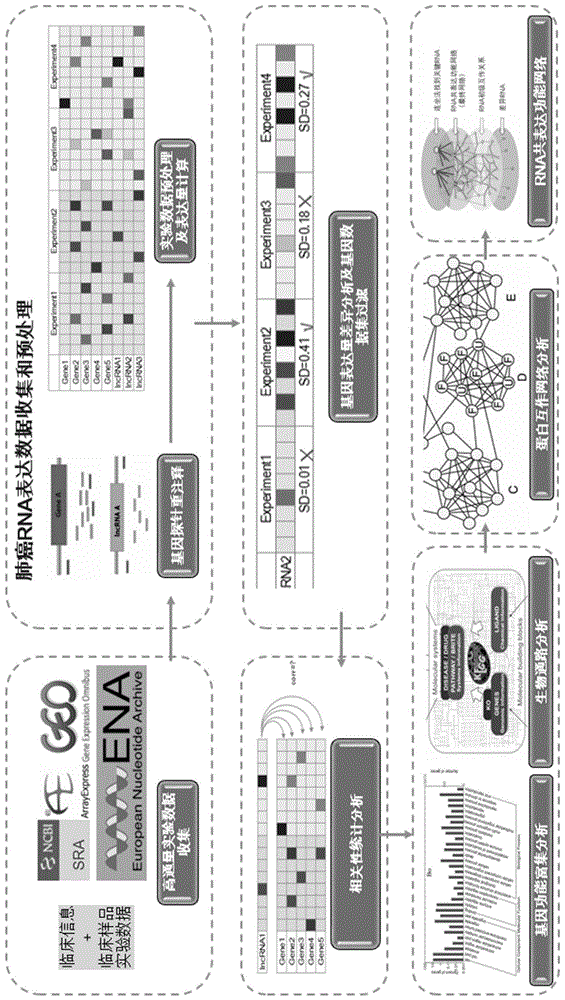
图1是rna共表达功能网络分析方法的基本步骤;
图2是本发明一些实例rna相关性矩阵建立方法的示意图;
图3是实施例1中rnaseq测序结果总结;
图4是实施例1中显著差异基因的表达量热图分析(红色为高表达,绿色为低表达);
图5是差异基因rp11-434d9.1的rna基因功能富集关系和生物通路富集评关系这两部分评分的计算结果;
图6是差异基因rp11-434d9.1的rna-蛋白互作关系网络;
图7是last1在以下细胞系的qpcr检测结果:a)last1过表达的a549细胞系;b)last1低表达的a549细胞系;
图8是last1表达量与病人生存时间关系的分析结果:左图:多维cox回归分析(x轴:生存时间,y轴:差异表达倍数)右图:用survivalr包对last1进行kaplan-meier(log-rank)分析;
图9是实施例2的差异基因火山图(logfc即log2foldchange,指示正常肺组织和肿瘤样品之间每个基因表达的倍数变化);
图10是上调差异rna进行表达量的关联分析;
图11是上调差异rna的基因功能富集权重分析和生物通路关系权重分析;
图12是下调差异rna进行表达量的关联分析;
图13是下调差异rna的基因功能富集权重分析和生物通路关系权重分析;
图14是6个重要的转录调控因子富集信息;
图15是分析得到的基因-蛋白互作调控网络。
具体实施方式
下面以肺癌为例,示意性说明基于肿瘤高通量实验数据进行关键rna功能挖掘的方法。
该方法在临床试验数据的基础上,对转录芯片探针进行重新注释,整合了多种蛋白质-蛋白质相互作用(ppi)网络信息,并在基因功能数据库和生物通路数据库的基础上,融合了转录因子分析,并且与各种肺癌基因数据库对接进行筛选,最终能找到对人体内的待挖掘功能的临床肺癌相关的关键rna,并起到功能和机制提示作用。
rna共表达功能网络分析方法的基本步骤如图1所示,具体包括:
1、高通量实验数据收集、预处理和表达量计算
rna差异表达分析可以建立在rna芯片或rna-seq获得的原始实验数据上进行。对于由rna芯片或rna-seq获得的原始实验数据,需要先进行以下原始信号处理:
1)对于由rna芯片获得的原始实验数据,需要先进行以下原始信号处理:
a)背景处理和数据清洗:先使用工具对rna芯片数据进行背景信号,过滤掉芯片杂交信号中属于非特异性的背景噪音部分。一般以图像处理软件对芯片划格后,每个杂交点周围区域各像素吸光度的平均值作为背景,但此法存在芯片不同区域背景扣减不均匀的缺点。也可利用芯片最低信号强度的点(代表非特异性的样本与探针结合值)或综合整个芯片非杂交点背景所得的平均吸光值作为背景。完成背景处理后,对于负值和噪声信号,使用变异系数法和k邻近法进行数据清洗。本步骤可使用的工具例如r软件的affy/limmapackage。
b)提取探针的表达值和探针表达值的归一化:经过背景处理和数据清洗处理后的修正值反映了基因表达的水平。然而在芯片试验中,各个芯片的绝对光密度值是不一样的,在比较各个试验结果之前必需将其归一化(normalization,也称作标准化),从而调整由于基因芯片技术引起的误差。本步骤可以采用平均数、中位数标准化方法进行归一化。
c)探针表达值转换为rna表达值(探针注释):传统芯片探针注释是使用芯片厂家提供的注释信息,或者使用r软件的bioconductor工具库进行注释,其原理都是使用芯片开发时候厂家定下来的探针序列和基因对应关系进行注释。本方法特意本步骤引入新的注释方法,希望不要漏掉可能重要的新rna。传统的芯片分析方法里面,每个探针都有确定的固定对应的基因名。但是随着rna数据库的更新,有很多未知探针,现在已经可以对应到新发现的rna数据库中。如果只是使用传统的分析方法,可能会漏掉很多重要的新rna。因此发明人重新整合了一个新的更加全面的rna数据库。
重新整合的rna数据库包括:
1)lncrnadb:提供有生物学功能的长链非编码rna的全面注释。这是长链非编码rna研究领域的权威johnmattick实验室构建的网站。
2)gencodehuman(https://www.gencodegenes.org/human/),
3)refseq
4)ensembl,ensemble数据库可为药物研发提供超过167000种生物活性化合物包括化学结构在内的必要信息。本数据库利用用户容易掌握的界面将数据、文本和图象资料有机地结合起来,便于查询。ensemble可从药品专利开始,再通过其临床前和临床研究资料,直至注册信息、市场概况及其他方面的相关资料来跟踪药物。数据库每月更新一次,每年增加约10000种新化合物。
5)lncatlas(https://lncatlas.crg.eu/)
6)noncode:noncode提供对长链非编码rna的全面注释,包括表达和ncfans计算机软件预测的lncrna功能。这是非编码rna研究的知名数据库
7)humanbodymaplincrnas,
8)lncrnome:超过18000转录本目前已作为lncrna标注,覆盖先前注释非编码转录本,包括大型基因间非编码rna,反义rna和加工的假基因。但在提供稳定的注释,交叉引用和生物相关的信息资源方面有显著的差距。由印度csir基因组和整合生物学研究所研究人员开发的lncrnome,旨在填补这一空白,他们通过把生物显著性的各种各样的信息注释整合到一个全面的知识库。
9)lugend:肺癌基因数据库(lungcancergenedatabase)是一个经过整理和集成的基于web的数据库,用于存储人类肺癌基因的基因和基因组位点。lugend涵盖了与肺癌的许多分子和遗传事件有关的基因,包括文献中发表的染色体位置,突变和表达。
同时,有些探针会于多个rna发生重叠,发明人也需要对此类探针的表达量进行评估。因此发明人通过芯片探针重注释策略,对每个芯片探针进行基因组定位,并把rna序列映射到基因组上,通过位置叠加的关系,与发明人重新整合的rna数据库进行交叠,找出rna所对应的芯片探针。
位置叠加转换rna表达值的原则如下:
i)如果一个探针只与一个rna转录本发生重叠,则rna转录本表达值=探针表达值;
ii)如果一个探针与两个以上rna转录本发生重叠,且所处的正负链方向一致,则rna转录本1表达值(1v2)=探针表达值,rna转录本2表达值(1v2)=探针表达值;
iii)如果一个rna转录本与两个以上探针发生重叠且基因组距离小于1000bp,则rna转录本表达值(2v1)=(探针1表达值+探针2表达值)*50%;
iv)如果rna转录本表达值同时存在1v2和2v1的情况,则需要计算综合的rna转录本表达值=rna转录本表达值(2v1)-rna转录本表达值(1v2),作为最终的rna转录本表达值。
v)但如果转换后的rna转录本表达值存在负值,则采用k邻近法进行数值校正。
2)对于由rna-seq获得的原始实验数据,需要先进行以下原始信号处理
a)去除测序数据中的接头,使用fastx_toolkit里面的fastx_clipper工具;
b)去除测序数据中的低质量reads,使用fastx_toolkit里面的fastq_quality_filter工具;
c)数据比对,使用tophat、bowtie、hisat2、bwa之类的工具都可以;
d)计算rna表达值,可以使用的工具有htseq-count或stringtie、bedtools、gfold等。
2、基因表达量差异分析及基因数据集过滤
根据研究目的或实验设计,可对多组样本两两之间进行,具体方法包括差异倍数分析、t检验等,获取差异rna列表。特别的,临床检测样本小于15pair的情况下,发明人认为临床检测样品数据量过少,可以在选出初步的差异基因后,加入类似的已发表的数据集(发明人以下称为伴随数据集),但是要先对数据集进行过滤。过滤的原则包括以下:1)与研究的临床特征相符;2)control/test的对照设计一致;3)根据临床数据集里rna表达量,找出初步差异rna,用这个初步差异rna的集合去计算候选已发表数据集中的相应rna的表达值标准差。如果标准差小于0.2,则认为初步筛选出来的差异rna在候选已发表数据集中不具备相似特性,不能入选伴随数据集,不能与临床检测样品一起进行分析。伴随数据集的选择来源:1)ncbigeo;2)ncbisra;3)ena–ensemblenucleotidearchive。这些来源的伴随数据集均有原始数据下载,确保可以与临床数据通过批间校正后进行联合分析。
3、建立rna相关性矩阵scor
计算rna探针与本数据集中其他任意探针(n个)的pearson相关系数,从而得到相关性矩阵。最后利用rankaggregation计算出rna探针与其他探针的相关性p-value,按照p-value数值从小到大排序,得到与rna探针所相关的其他探针的重要性排序。按照相同的方法,计算k个实验里面的相关性排序,然后综合k个实验的各自结果得到每个探针相关性排序的总排名(图2)。
4、获得共表达关系权重评分sco
根据p-value显著性阈值(p<0.05),提取显著差异rna的最相关的探针列表,并找出相应探针所对应的基因,即得到与rna相关的共表达基因。相关性越大,sco越高分。
5、进行rna的基因功能富集权重评分sgo
进行计算的基因功能数据库为geneontology(http://geneontology.org/),使用r软件中的clusterprofiler包进行分析,也可使用metascape(http://metascape.org/)和string数据库(https://string-db.org/)进行在线分析。p值越显著,sgo越高分。
6、进行rna的生物通路关系权重评分skegg
进行计算的生物通路数据库为kegg(京都基因与基因组百科全书,kyotoencyclopediaofgenesandgenomes,www.kegg.jp)。kegg(京都基因和基因组百科全书)是了解高级功能和生物系统(如细胞、生物和生态系统),从分子水平信息,尤其是大型分子数据集生成的基因组测序和其他高通量实验技术的实用程序数据库资源,由日本京都大学生物信息学中心的kanehisa实验室于1995年建立,是国际最常用的生物信息数据库之一,以“理解生物系统的高级功能和实用程序资源库”著称。本步骤使用r软件中的clusterprofiler包进行分析,也可使用david(https://david.ncifcrf.gov)和kobas数据库(http://kobas.cbi.pku.edu.cn)进行在线分析。p值越显著,skegg越高分。
7、进行rna的蛋白互作关系权重评分sppi
本步骤进行计算的蛋白互作数据库是整合多种开放的蛋白互作数据库所建立的。多个数据库中的数据进行合并去冗余,并把多个数据库的综合评分相乘得到最终sppi。整合的数据库包括以下:
1)igdb.nsclc数据库:非小细胞肺癌综合基因组数据库(integratedgenomicdatabaseofnon-smallcelllungcarcinoma)旨在促进和确定已鉴定的肺癌基因和microrna的优先级,以进行肺肿瘤发生的病理学和机制研究以及开发新的临床干预策略。
2)scop数据库:蛋白质结构分类数据库(structuralclassificationofprotein,scop)是对已知蛋白质结构进行分类的数据库,根据不同蛋白质的氨基酸组成以及三级结构的相似性,描述已知结构蛋白质的功能及进化关系。scop数据库的构建除了使用计算机程序外,主要依赖人工验证。
3)dip数据库:蛋白相互作用数据库(databaseofinteractingprotein,dip)研究生物反应机制的重要工具,收集了经实验验证的来自文献报道的蛋白质相互作用。数据库包括蛋白质的信息、相互作用的信息和检测相互作用的实验技术三个部分。用户可以根据蛋白质、生物物种、蛋白质超家族、关键词、实验技术或引用文献来查询dip数据库。
4)string数据库:string数据库是一个搜索已知蛋白质之间和预测蛋白质之间相互作用的数据库,该数据库可应用于2031个物种,包含960万种蛋白和1380万中蛋白质之间的相互作用。它除了包含有实验数据、从pubmed摘要中文本挖掘的结果和综合其他数据库数据外,还有利用生物信息学的方法预测的结果。
5)spike数据库:spike(signalingintegratedknowledgeengine)主要存储蛋白质在生物信号通路中的相互作用关系以及大量的生物信号通路间关联互作信息。里面包含的信息主要偏向于dna损伤、细胞周期、模式生物死亡以及相关通路。
6)reactome数据库:reactome是一个汇集了由专家撰写,经同行评阅的有关人体内各项反应及生物学路径的文章的数据库。该数据库为人们提供了一个全新的从整体水平上对生物学途径进行研究的工具,提供了直观的生物信息学工具,用于可视化,解释和分析通路相关知识,以支持基础研究,基因组分析,建模,系统生物学研究等。
7)pfam数据库:pfam数据库是蛋白质家族的数据库,根据多序列比对结果和隐马尔可夫模型,将蛋白质分为不同的家族。
8)pdb数据库:pdb蛋白质结构数据库(proteindatabank,简称pdb),是目前最主要的收集生物大分子(蛋白质、核酸和糖)2.5维(以二维的形式表示三维的数据)结构的数据库,是通过x射线单晶衍射、核磁共振、电子衍射等实验手段确定的蛋白质、多糖、核酸、病毒等生物大分子的三维结构数据库。
9)mint数据库:mint(molecularinteractiondatabase)是一个蛋白质相互作用的数据库,该数据库中的蛋白相互作用都是由专家审核过的有实验证据支持的,目前该数据库涵盖了607个物种,共117001个蛋白相互作用关系。
10)intact数据库:intact数据库是一个存储和分析生物分子间相互作用的公共数据库,主要记录蛋白质相互作用及试验方法、实验条件和相互作用数据库,数据主要来自文献的人工检验或用户提交。
11)hprd数据库:hprd,全名是人类蛋白质参考数据库(humanproteinreferencedatabase)。该数据库是目前最大的人类蛋白相互作用数据库,包含30000多个蛋白质和41000对条相互作用信息。除了包含蛋白相互作用信息,hprd还囊括了蛋白注释、亚细胞定位、结构域、转录后修饰和信号通路合集等多种功能。
12)biogrid数据库:生物通用交互数据集库(biogrid)是一个公共数据库,用于存档和传播来自模型生物和人类的遗传和蛋白质相互作用数据。biogrid目前拥有超过1500000种来自高通量数据集和个人重点研究的互动,这些研究来自于初级文献中的63000多份出版物。
8、疾病-rna关系评分sdisease:用到的数据库涉及以下。在不同数据库出现的次数越多,在同一个疾病描述中共同出现的rna,其疾病关联评分越高。
1)lncrnadisease(http://cmbi.bjmu.edu.cn/lncrnadisease),
2)omim
3)ncbiclinvar数据库
4)hlungdb:非小细胞肺癌综合基因组数据库(integratedgenomicdatabaseofnon-smallcelllungcarcinoma)旨在促进和确定已鉴定的肺癌基因和microrna的优先级,以进行肺肿瘤发生的病理学和机制研究以及开发新的临床干预策略。
9、调控因子tf关系评分stf
进行rna的cis或trans的targets预测及rna上游的tf预测,cis表示染色体邻近位置的靶点;trans表示不同染色体或染色体远端位置的靶点。只需明确rna共表达基因在染色体上的远近位置,即可找出cis或trans的靶点。另外对rna可以进行chip-seq信号检测,从而预测上游tf信息。因此,对于rna,发明人亦可类似构建tf-rna的调控网络。另外还可以用到transcfac转录因子数据库。用到的数据库有starbase(http://starbase.sysu.edu.cn/)、chipbase(http://deepbase.sysu.edu.cn/chipbase/)等。
10、关系综合评分score
根据上述各步骤得到的关系分数,按照公式score=scor*sco*sgo*skegg*sppi*sdisease*stf计算任意两个rna相互关系的综合评分。以上关系评分,并非任意两个rna之间都一定有数据库结果支持,对于没有数据库结果支持的两个rna,其相应的分值定为1。根据分值高低,可以定位到本次临床指征相关的重要rna。分值越高,是本次实验数据临床指征相关的可能性越高。最终根据连坐法形成最终的rna共表达功能网络。
下面结合实例,进一步说明本发明的技术方案。
实施例1:
1)样品收集:以临床上已经确诊病理并能获得石蜡组织的肺腺癌患者为研究对象,入组病人100例。基于高通量转录组测序(rna-seq)方法分别对组织样本(癌和癌旁,共200个样品)进行转录组测序和生物信息分析。研究对象均选自如thoraciccancer9(2018)1680–1686的表1所示,样品筛选要求如下:肺腺癌,有确诊信息,有完整临床随访信息。
2)rna测序数据预处理:使用fastx_toolkit里面的fastx_clipper工具去除测序接头,使用fastx_toolkit里面的fastq_quality_filter工具去除低质量的测序reads,然后使用tophat进行数据比对,参考基因组为humanhg19。从而计算得到每个样品的原始测序reads和比对上的测序reads数,测序原始数据预处理结果总结如图3,从图中可知:每个样品的平均测序量>12mreads,比对上的reads>8.5mreads,比对率>70%。临床数据>15pairs,因此不需要选择伴随数据集。
3)差异基因分析和共表达关系权重分析:使用htseq-count计算rna表达量,对定量后的rna进行差异比较分析,对显著差异基因(p<0.01)绘制表达关系图如图4(红色为高表达,绿色为低表达)。
4)本次分析中用到的评分标准包括:rna基因功能富集关系评分、生物通路富集评分、蛋白互作关系评分。下面是以其中一个差异基因rp11-434d9.1为例,展示rna基因功能富集关系和生物通路富集评关系这两部分评分的计算结果(图5)。
5)结合蛋白互作关系数据库,最终形成网络如图6。图中核心是rp11-434d9.1,红色是上调表达的rna(与核心rna一起,是最重要值得关注的rna),绿色是次级关联rna(较为重要),蓝色是三级关联rna(相对不重要)。
6)表达量与临床指征的关联分析:为了验证得到的最重要的rp11-434d9.1核心rna(命名为last1),发明人进行了过表达和低表达的细胞实验(图7),并使用cox分析,按照表达量的中位值将病人划分为高表达组和低表达组。然后使用乘积极限法来估计生存率及使用kaplan-meier(km)法估计和绘制生存曲线,并使用log-rank检验进行高低表达组生存曲线的比较,经过比较高低表达组两者差异显著,提示last1表达量的差异能预测病人的生存时间(图8),并且。与本次实验数据的临床预期吻合。证实使用本套分析流程得到的关键核心rna,是可靠,符合临床的。
实施例2
1)样品收集:以临床上已经确诊病理并能获得石蜡组织的小细胞肺癌患者为研究对象,入组病人10例。基于rna高通量测序方法分别对组织样本(肺癌组织及癌旁组织,共20个样品)进行检测。样品筛选要求如下:小细胞肺癌,所有患者标本均经病理科确诊,术后生存时间均超过3个月,有完整临床随访信息。
2)伴随数据集的选取:利用公共数据库资源,从美国国立生物技术信息中心(nationalcenterforbiotechnologyinformation,ncbi)的基因表达数据库(geneexpressionomnibus,geo)下载了86个小细胞肺癌样本的rna-seq原始数据,数据集编号是gse60052,下载链接为https://www.ncbi.nlm.nih.gov/sra?linkname=bioproject_sra_all&from_uid=257389,这些样本的临床信息与本分析的临床样品吻合,可以合并分析。
3)数据预处理:本次分析用到的软件为subread,samtools,bamtools,参数为默认值。
4)rna表达量计算:发明人使用htseq-counts软件分析了rna-seq数据的原始计数,bedtools和counts软件featurecounts以计算表达值。
5)差异rna分析:使用bioconductor中的edger软件包进行差异表达分析。筛选基因表达差异的参数:padj<0.01和log2foldchange>0,位居前500位。图9火山图展示了总体差异rna的情况,红色和绿色部分的点就代表值得关注的差异rna。
6)上调差异rna的共表达矩阵分析、基因功能富集分析和生物通路富集分析。
a)根据logfc进行排序,logfc越大且pvalue越小,则差异越显著。以下展示前100个显著差异的上调差异rna。
b)共表达关系权重分析:根据差异rna筛选原则,对前500个显著差异的上调差异rna进行表达量的关联分析(图10)。相关性越强的差异rna,距离越近。红色代表表达量高,蓝色代表表达量低。
c)上调差异rna的基因功能富集权重分析和生物通路关系权重分析(图11)7)下调差异rna的共表达矩阵分析、基因功能富集分析和生物通路富集分析。
a)根据logfc进行排序,logfc越小且pvalue越小,则下调差异越显著。以下展示前100个显著差异的下调差异rna。
b)共表达关系权重分析:根据差异rna筛选原则,对前500个显著差异的下调差异rna进行表达量的关联分析(图12)。相关性越强的差异rna,距离越近。红色代表表达量高,蓝色代表表达量低。
c)下调差异rna的基因功能富集权重分析和生物通路关系权重分析(图13)。
8)调控因子tf关系分析:对上调和下调的显著差异rna均进行tf关系分析,使用到的数据库是transcfac转录因子数据库,分析得到6个重要的转录调控因子富集信息(图14)。
9)疾病-rna关系权重分析:本步骤分析发现ezh2、tacc3在多个肺癌数据库中出现,出现频次为7。
10)综合以上结果筛选出细胞周期和神经系统发育这2条(同时也是“p-值”最显著的)通路的rna基因进行深入研究,由此发现:
a)细胞周期通路异常激活:细胞周期通路研究中发现,基因cdc7、e2f1、cdc6、cdk1、e2f2、pkmyt1、cdc20、espl1、mcm4、cdc25a、ccne2、cdkn2a、plk1、cdkn2c、bub1、orc6、orc1、brsk1等82个基因显著富集在kegg细胞周期通路。
b)神经系统发育通路异常激活:神经系统发育通路研究中发现,prox1、aspm、drd2、sox4、e2f1、brsk1、insm1、cdk5r1、phgdh、ush1c、fzd3、tacc3、stmn1、aurka、stil、kif14、dnmt3a、ezh2等92个基因显著富集在kegg细胞周期通路。
11)把共表达相关性结果、基因功能富集、生物通路富集、调控因子tf关系等结合分析,发明人发现两条通路有21个共同基因,其中15个基因聚集在同一个蛋白互作调控网络。
12)恶性肿瘤最基本的生物学特征之一是细胞周期调控紊乱导致的细胞恶性转化和肿瘤细胞失控性增殖。了解细胞周期的调控机制能够揭示肿瘤发生发展的异常情况,阐释癌症的发生机制,从而为肿瘤的早期诊断和临床治疗提供分子标志物及药物靶点。其次,小细胞肺癌是一种神经内分泌肿瘤,其发生发展与神经系统功能异常密切相关。上述网络核心rna均与神经系统密切相关。
实施例2中,临床样品数量较少,使用本分析方法,查询到伴随数据集,包括86个可一起用于该案例同步分析的数据。这种加入类似的伴随数据集的方法,使用明确的过滤原则,使得初步筛选出来的差异rna在候选伴随数据集中具备相似特性,从而增加了临床研究的统计意义。
以上是对本发明所作的进一步详细说明,不可视为对本发明的具体实施的局限。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的简单推演或替换,都在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!