一种复方紫草微乳温敏原位凝胶及其制备方法和应用
本发明涉及药物制剂领域,具体涉及一种复方紫草微乳温敏原位凝胶及其制备方法和应用。
背景技术:
复方紫草油具有清热凉血,解毒止痛等功效,除可抗菌消炎外还有生肌和促进上皮生长的功能,其止痛效果好,创面愈合快,用于妇科阴道炎、宫颈糜烂等疾病治愈率高,疗效确切,阴道不适症状可得到明显改善,且不影响宫颈的弹性,具有良好的临床应用前景。
专利cn112426455a公开了一种紫草油组方,由以下重量份的药材制备得到:紫草54-66份;白芷32.4-39.6份;当归18-22份;地榆10.8-13.2份;黄柏10.8-13.2份;冰片10.8-13.2份;胡麻油900-1100份;通过本紫草油应用于制备紫草油纱条具体的制备过程,能够得到药液均匀、稳定的紫草油纱条,提高了紫草油中左旋紫草素的含量,具有使用方便、创面的修复作用显著的优点。
复方紫草油在临床的使用方法为:将油制剂制备成含药纱条,将纱条填入患者阴道内,使其紧贴宫颈糜烂面,30min后取出或留出尾端于阴道口外,嘱患者睡前取出。存在患者自主给药困难、使用时有异物感、给药次数频繁、患者顺应性差等缺点,因此有必要将其制备成阴道黏膜给药制剂。
贾颖等公开了一种复方紫草凝胶外用制剂(《日用化学工业》,第50卷第12期,2020年12月);周彤等公开了一种复方紫草凝胶剂(复方紫草凝胶剂的制备研究,天津工业大学硕士论文);cn110292591a公开了一种复方紫草凝胶及其制备方法与应用。但现有技术的凝胶剂在常规下即为半固体状态,仍不适用于阴道给药。
温度敏感型原位凝胶是一类以溶液状态给药后,在生理条件下,转化为非化学交联的半固体制剂,制备基质常选用p407和p188。其低温呈低黏度流体状态,置入阴道后,在体温条件下,与多皱褶的阴道黏膜组织紧密贴合,并快速转变为半固体凝胶,可增强药物生物粘附性、延长药物滞留时间并提高其生物利用度。
技术实现要素:
针对现有技术存在的问题,本发明提供了一种复方紫草微乳温敏原位凝胶,并对其性状、粒径、流变学性质、体外释放度等进行了研究。所述复方紫草微乳温敏原位凝胶在室温条件下为液体状态,以液态进行阴道给药,在体温下凝胶黏度迅速增大转变为半固体状态,并与阴道黏膜紧密结合,提高药物的滞留时间;并且室温下以微乳形式存在,活性成分有较高的溶解度,有效降低辅料的添加量,从而减小制剂的刺激性。
本发明是通过以下技术方案来实现的。
一种复方紫草微乳温敏原位凝胶,其特征在于,所述原位凝胶的组分包括复方紫草药物干浸膏、油相、乳化剂、助乳化剂和温敏凝胶基质。
进一步的,所述复方紫草微乳温敏原位凝胶中各组分的重量配比为:复方紫草干浸膏1-3份、油相2-6份、乳化剂4.5-13.5份、助乳化剂1.5-4.5份、温敏凝胶基质10-25份、水50-100份。优选为复方紫草干浸膏2份、油相4份、乳化剂9份、助乳化剂3份、温敏凝胶基质12份、水82份。
进一步的,所述复方紫草药物干浸膏的原料组成包括紫草、当归、地榆、白芷和冰片,重量配比为紫草20-40份∶当归5-15份∶地榆1-10份∶黄柏1-10份:白芷12-24份∶冰片1-10份;优选为紫草30份∶当归10份∶地榆6份∶黄柏6份:白芷18份∶冰片6份。
进一步的,所述复方紫草药物干浸膏的制备方法为称取处方量的紫草、当归、地榆、白芷、冰片,加入乙醇,超声提取,滤去药渣,醇提液挥干溶剂,得药物干浸膏。
进一步的,所述油相选自丙二醇辛酸酯(capryol)、肉豆蔻酸异丙酯(ipm)、油酸甘油酯(peceol)、油酸乙酯、辛癸酸甘油酯、辛酸甘油三脂、亚油酸乙酯、辛酸乙酯、丁酸乙酯、鲸蜡醇十六酸酯、薄荷油和大豆油中的一种或几种的混合。所述油相优选肉豆蔻酸异丙酯(ipm)。
进一步的,所述乳化剂选自司盘80、吐温-40、吐温-80、聚氧乙烯硬脂酸酯、聚氧乙烯脂肪酸酯、聚氧乙烯(20)油醚、壬基酚聚氧乙烯醚、辛酸癸酸聚乙二醇甘油酯(labrasol)、聚氧乙烯蓖麻油、聚氧乙烯氢化蓖麻油rh40(cremophorrh40)、聚氧乙烯氢化蓖麻油el-p(cremophorel-p)、聚氧乙烯(35)蓖麻油(kolliphorelp)、泊洛沙姆和大豆磷脂中的一种或几种的混合。所述乳化剂优选聚氧乙烯(35)蓖麻油(kolliphorelp)。
进一步的,所述助乳化剂选自无水乙醇、异丙醇、丙三醇、1,2-丙二醇、乙二醇单乙基醚(transcutolp)、聚乙二醇、二乙二醇单甲醚中的一种或几种的混合。所述助乳化剂优选丙三醇。
进一步的,所述温敏凝胶基质选自泊洛沙姆、壳聚糖、聚n-异丙基丙烯酰胺中的一种或几种的混合。所述温敏凝胶基质优选由泊洛沙姆p407和泊洛沙姆p188组成;其中,泊洛沙姆p407与泊洛沙姆p188质量配比为(10-20)∶(2-4),优选为10∶2。
进一步的,所述复方紫草微乳温敏原位凝胶中各组分的重量配比为:复方紫草干浸膏1-3份、ipm2-6份、聚氧乙烯(35)蓖麻油4.5-13.5份、丙三醇1.5-4.5份、p40710-20份、p1882-4份、水50-100份;优选为复方紫草干浸膏2份、ipm4份、聚氧乙烯(35)蓖麻油9份、丙三醇3份、p40710份、p1882份、水82份。
另一方面,本发明还提供了所述复方紫草微乳温敏原位凝胶的制备方法,其特征在于,包括如下步骤:
(1)复方紫草药物干浸膏的制备;
(2)称取处方量的复方紫草干浸膏,置油相中超声溶解;取处方量乳化剂和助乳化剂加入上述油相中,混合均匀,加超纯水使成澄清透明液体,制成复方紫草微乳;
(3)向上述复方紫草微乳中加入处方量温敏凝胶基质,采用冷溶法制备复方紫草微乳温敏原位凝胶。
进一步的,所述步骤(1)为称取处方量的紫草、当归、地榆、白芷、冰片,加入药材4-8倍量95%乙醇,超声提取,滤去药渣,醇提液挥干溶剂,得药物干浸膏。进一步优选为称取处方量的紫草(粉碎或直径小于1cm细块)30g、当归10g、地榆6g、白芷18g、冰片6g,加入药材6倍量95%乙醇,40℃超声40min提取,滤去药渣,醇提液于60℃水浴挥干溶剂,得药物干浸膏;干浸膏中左旋紫草素含量为1.72mg/g。
进一步的,所述步骤(2)为称取复方紫草干浸膏2.0g,置4.0gipm中超声加速溶解,取混合乳化剂[聚氧乙烯(35)蓖麻油9.0g和丙三醇3.0g)]加入上述油相中,室温下磁力搅拌混合均匀,缓慢滴加超纯水至100.0g,使成澄清透明液体,制成微乳。
进一步的,所述步骤(3)为向上述复方紫草微乳中加入处方量温敏凝胶基质p407和p188,充分搅拌后置于4℃冰箱溶胀24h,取出该微乳-原位凝胶液,缓慢搅拌使分散均匀,使用稀盐酸将凝胶液ph调节至4.0~5.5范围内,即得复方紫草微乳-原位凝胶液。
第三方面,本发明还提供了所述复方紫草微乳温敏原位凝胶在制备治疗阴道炎和/或宫颈糜烂药物中的应用。
本发明的有益效果:
1、复方紫草微乳温敏原位凝胶在室温条件下为液体状态,以液态进行阴道给药,溶液状态的凝胶可迅速在褶皱状的阴道黏膜表面铺展,覆盖完全;由于温度的变化,在体温下凝胶黏度迅速增大、强度急剧提高,进而转变为半固体状态并与阴道黏膜紧密结合,可提高药物的滞留时间,更好的发挥疗效。
2、复方紫草微乳温敏原位凝胶中活性成分在微乳处方辅料中有较高的溶解度,可有效降低辅料的添加量,从而减小制剂的刺激性。因此,通过测定药物干浸膏在油相、乳化剂和助乳化剂中的饱和溶解度,选择ipm、聚氧乙烯(35)蓖麻油和丙三醇分别作为油相、乳化剂和助乳化剂。
附图说明
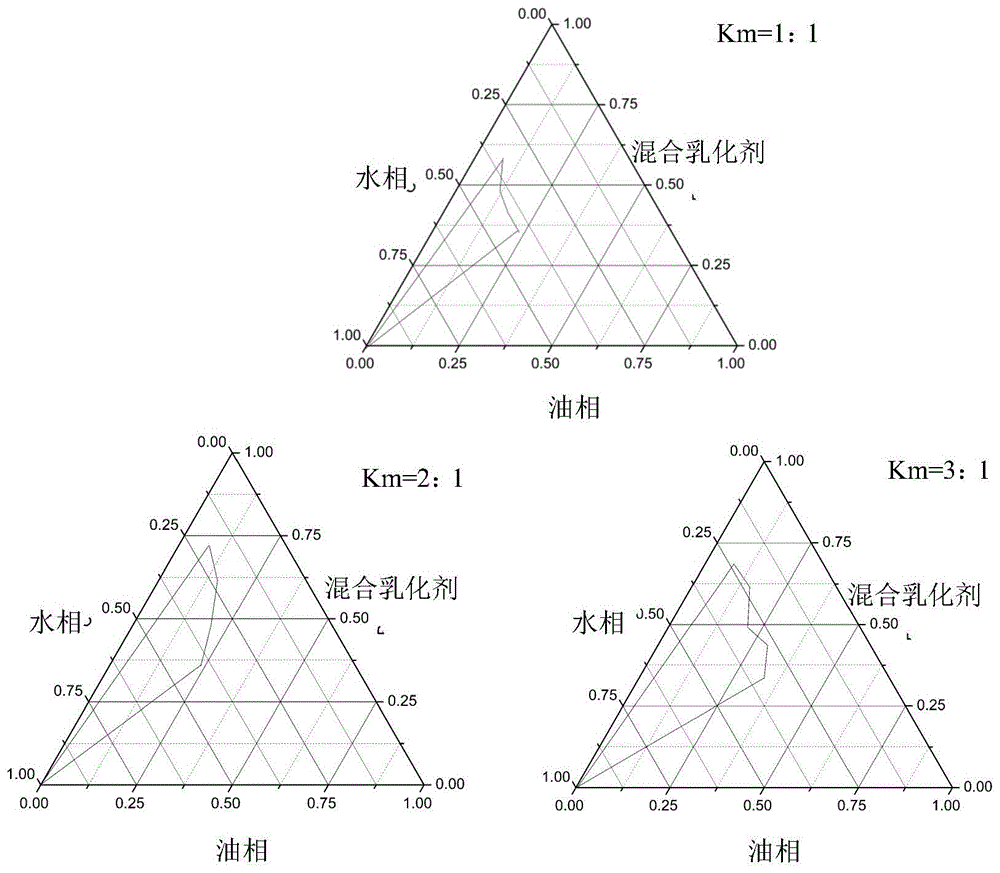
图1是不同乳化剂和助乳化剂比例微乳的伪三元相图。
图2是左旋紫草素高效液相色谱图,其中a为空白凝胶;b为左旋紫草素对照品;c为复方紫草微乳-原位凝胶。
图3是复方紫草微乳(a)和微乳-原位凝胶胶凝前后(b、c)的外观图。
图4是复方紫草微乳(a)和微乳-原位凝胶(b)的粒径分布图。
图5是复方紫草微乳(a)、未胶凝(b)与胶凝(c)微乳-原位凝胶的透射电镜扫描图。
图6是复方紫草微乳-原位凝胶累积溶蚀曲线。
图7是复方紫草微乳-原位凝胶累积释放曲线。
具体实施方式
下面将通过实施例来进一步解释和说明本发明的技术方案。应当理解为:本发明的实施例仅为说明本发明而给出,并非对本发明的限制,在本发明技术方案的前提下任何对本发明的简单改进均属于本发明的保护范围。
本发明涉及的仪器包括但不限于:高效液相色谱仪(waters2489紫外检测器,1525泵);ms105du型分析天平(上海梅特勒-托利多仪器有限公司,感量:0.01mg);thz-d恒温振荡器(苏州培英实验设备有限公司);tgl-16c台式离心机(上海安亭科学仪器厂);79-1磁力加热搅拌器(江苏江阴科研器械厂);st2100ph计(常州奥豪斯仪器有限公司);malvernnanozs-90型粒度电位仪(英国马尔文仪器有限公司);h-7650透射电子显微镜(日本日立)。
本发明涉及的试剂包括但不限于:左旋紫草素对照品(中国食品药品检定研究院,批号:110769-200506,含量:99.8%);泊洛沙姆407(德国basf公司,批号gnd11321b)、泊洛沙姆188(德国basf公司,批号gnd10221b);聚氧乙烯(35)蓖麻油(德国basf公司,批号95284497v0);肉豆蔻酸异丙酯(ipm,德国basf公司,批号00018999798);丙三醇(天津市大茂化学试剂厂,批号:20190401);磷酸(天津市大茂化学试剂厂,批号20170801);甲醇(美国fisherchemical公司,色谱纯,批号190291)。
实施例1复方紫草药物干浸膏的制备
称取处方量的紫草(粉碎或直径小于1cm细块)30g、当归10g、地榆6g、白芷18g、冰片6g,加入药材6倍量95%乙醇,40℃超声40min提取,滤去药渣,醇提液于60℃水浴挥干溶剂,得药物干浸膏。干浸膏中左旋紫草素含量为1.72mg/g。
实施例2药物干浸膏在各辅料中的溶解度测定
测定复方紫草药物干浸膏在不同辅料(油相、乳化剂和助乳化剂)中的饱和溶解度。将足量的干浸膏分别加入含有2ml不同溶媒的试管中,置涡旋振荡器涡旋10min,使充分混匀,置37℃±1℃恒温振荡器振荡72h,使药物充分溶解;然后,将混合物5000r/min,离心15min,以0.45μm微孔滤膜过滤;取续滤液,采用hplc法分析其质量浓度,计算溶解度。
药物干浸膏在不同溶媒中的溶解度测定结果,见表1。结果表明,药物浸膏在油相ipm、乳化剂聚氧乙烯(35)蓖麻油和助乳化剂丙三醇中的溶解度最大。故选择ipm、聚氧乙烯(35)蓖麻油和丙三醇分别作为油相、乳化剂和助乳化剂,通过绘制伪三元相图确定微乳各相比例。
表1药物干浸膏在不同溶媒中的溶解度测得结果(n=3),
实施例3微乳各相比例的确定
根据溶解度测定结果,选择ipm为油相,聚氧乙烯(35)蓖麻油为乳化剂,丙三醇为助乳化剂。表面活性剂与助表面活性剂分别以质量比(即km值)3︰1、2︰1、1︰1混合均匀,制成混合溶液。将混合溶液与油相按照质量比9︰1、8︰2、7︰3、6︰4、5︰5、4︰6、3︰7、2︰8、1︰9的比例混匀,在持续磁力搅拌条件下,以超纯水缓慢滴加,直至体系平衡形成澄清透明的微乳,记录此时加入的水量。计算临界点油相、混合表面活性剂及水相在微乳体系中的质量分数,借助origin8.5软件绘制微乳的伪三元相图,并选择适宜的km值。
复方紫草微乳的伪三元相图如图1所示。当km值为3:1时,微乳区域面积较大,从相图中选择微乳的组成为油相(12.5%,w/w)、乳化剂系统(37.5%,w/w)和纯化水(50%,w/w),并确定km值为3∶1。
实施例4复方紫草微乳的制备
称取复方紫草干浸膏2.0g,置4.0gipm中超声加速溶解,取混合乳化剂[聚氧乙烯(35)蓖麻油9.0g和丙三醇3.0g)]加入上述油相中,室温下磁力搅拌混合均匀,缓慢滴加超纯水至100.0g,使成澄清透明液体,制成微乳。依此重复制备多批微乳液。
实施例5微乳-原位凝胶处方的确定
健康女性阴道内温度为37.2~37.8℃,理想的温敏凝胶应在尚未达到体温条件下即迅速成胶。因此,期望本制剂胶凝温度为34℃左右。p407溶液在达到临界胶束浓度和温度后,亲水性peo段和疏水性ppo段相互作用,紧密堆砌缠绕形成半固体凝胶,在p407中加入适量p188,通过调节peo/ppo的比例,达到理想的胶凝温度,使凝胶液能够在30℃内保持液态,且经模拟阴道液稀释后的凝胶在34℃左右时,形成半固体态凝胶。由于微乳中含有表面活性剂和油相,影响分子的物理聚集和缠结,达到理想胶凝温度时,凝胶基质的浓度显著降低。当p407和p188的浓度分别为100mg·ml-1和20mg·ml-1时,制备的微乳凝胶经模拟阴道液稀释后,胶凝温度为34.5℃。
采用冷溶法制备复方紫草阴道用微乳-原位凝胶。取制备好的微乳,加入处方量的p407和p188充分搅拌后置于4℃冰箱溶胀24h,取出该微乳-原位凝胶液,缓慢搅拌使分散均匀,阴道用制剂的ph值在4.0~5.5较适宜,故使用稀盐酸将凝胶液ph调节至该范围内,即得复方紫草微乳-原位凝胶液。
实施例6凝胶含量测定方法的建立
1、色谱条件
色谱柱:hypersilbdsc18(4.6mm×250mm,5μm);流动相:甲醇-0.1%磷酸(85∶15v/v);检测波长:516nm;流速:1ml·min-1;柱温:30℃;进样量:20μl。
2、供试品及对照品溶液的制备与测定
取复方紫草微乳-原位凝胶,精密移取5.0g于25ml量瓶中,加入适量甲醇,超声10min,使破乳,冷却至室温,甲醇定容,取溶液适量至离心管中,5000r·min-1离心15min取上清液,经0.45μm微孔滤膜过滤,取续滤液,作为供试品溶液。以左旋紫草素为对照品,精密称取对照品10mg于100ml量瓶中,加适量甲醇使药物溶解并定容,摇匀,作为储备液。精密移取1ml于10ml量瓶中,加甲醇稀释并定容,作为对照品溶液。按第1项中色谱条件进行测定,记录峰面积,以外标法计算含量。
3、方法学验证
专属性考察:分别取空白凝胶溶液、对照品溶液和供试品溶液按第1项中色谱条件进样,记录色谱图。空白凝胶、左旋紫草素对照品溶液、复方紫草微乳-原位凝胶的色谱图见图2。结果表明,左旋紫草素与杂质峰分离良好,杂质峰对测定结果无干扰。该方法准确可靠,适用于药物含量测定及释放行为的考察。
线性范围:精密配制系列浓度为2~30μg·ml-1的左旋紫草素甲醇溶液,作为标准溶液。按照第1项中色谱条件进行测定,记录峰面积。以左旋紫草素峰面积(a)为纵坐标,浓度(c)为横坐标,绘制标准曲线,并进行线性回归得标准曲线方程。配制系列浓度左旋紫草素对照品溶液,进行hplc分析,以峰面积(a)对浓度(c)进行线性回归,得回归方程a=29617c-2941.2,r=0.9999。结果表明,左旋紫草素在2~30μg·ml-1浓度范围内,浓度与峰面积呈良好的线性关系。
精密度:取浓度为10μg·ml-1的左旋紫草素溶液,按照第1项中色谱条件连续测定6次,记录峰面积,考察方法精密度。实验结果表明,左旋紫草素对照品溶液,重复进样的rsd为0.83%(n=6),表明该方法精密度良好。
回收率试验:精密称定已知含量的凝胶样品6份,分别加入一定量的左旋紫草素对照品溶液,按第2项中供试品处理方法处理上述样品,依第1项中色谱条件测定峰面积,并计算回收率。平均回收率为98.84%,rsd为1.14%,表明该方法准确度良好。
溶液稳定性:按照第2项中方法配制供试品溶液,室温下放置,分别于0h、2h、4h、6h、8h进样,记录峰面积,并求算rsd,考察样品溶液稳定性。实验结果表明,供试品溶液室温放置8h内,左旋紫草素含量无显著变化,rsd为1.37%。
在复方紫草微乳-原位凝胶的最佳处方与工艺条件下,分别制备3批(批号为:200615,200616,200618)样品,按照含量测定方法,每批样品测定3次,计算其平均含量。3批凝胶中左旋紫草素的含量分别为75.3,73.52和74.69μg·ml-1,rsd为1.21%(n=3)。
实施例7复方紫草微乳-原位凝胶的理化性质评价
1、外观性状
按照微乳和微乳-原位凝胶的优化条件,分别制备复方紫草微乳和微乳-原位凝胶,并对微乳、未胶凝的凝胶液与胶凝后的凝胶的外观形态进行观察。
复方紫草微乳液为澄清透明的紫红色溶液,流动性良好,经10000r·min-1离心10min后稳定不分层;微乳-原位凝胶在25℃时为紫红色且具一定流动性的液体;34℃胶凝后微乳-原位凝胶呈紫红色半固体态(图3)。
2、微乳离心稳定性
取复方紫草微乳5ml于离心管中,以10000r·min-1的转速离心10min,观察微乳性状。复方紫草微乳和微乳-原位凝胶,测得其平均粒径分别为(21.95±0.64)nm和(23.23±0.29)nm(n=3),粒径分布指数分别为(0.119±0.97)和(0.190±0.82)(n=3),其粒径分布较集中,且凝胶基质的加入对微乳粒径影响较小(图4)。微乳的zeta电位为(-14.44±2.64)mv,表明微乳较稳定。
3、粒径和形态测定
取复方紫草微乳和微乳-原位凝胶液,经适量纯化水稀释后于malvernnanozs-90型粒度电位仪平行测定3次,计算平均粒径和粒径分布指数,并测定其zeta电位。采用透射电镜观察微乳和微乳-原位凝胶在胶凝前后的形态。
利用透射电镜观察复方紫草微乳和微乳-原位凝胶发生胶凝化前后的形态特征(图5),由图可知,微乳及胶凝前后的凝胶中,乳滴均呈规则均匀的类球形,粒径较小,且分散性良好,无粘连。
4、黏度和流变学特性考察
将复方紫草微乳凝胶液与模拟阴道液按40:7.5混合,采用流变仪,测定非生理条件(25±0.1)℃和生理条件(35±0.1)℃下凝胶剂在不同剪切速率下对应的表观黏度值。
复方紫草微乳-原位凝胶经模拟阴道液稀释后的胶凝温度为34.5℃。在非生理条件下,凝胶液黏度值很小,且随剪切速率变化不大;而在模拟生理条件下形成凝胶后,凝胶呈剪切速率依赖性,随着剪切速率的增大,表现出剪切变稀的假塑性,呈现非牛顿流体特性。
5、凝胶的体外释放研究
采用无膜溶出法,将复方紫草微乳原位凝胶10ml加入已称重的平底带塞西林瓶中,置于37℃恒温振荡器中,待其完全胶凝后,加入37℃预热的模拟阴道液5ml,作为释放介质,于100r·min-1转速下振荡,每30min后,立刻倾出全部释放介质,并迅速称量此时西林瓶重量,然后将西林瓶重新放入振荡器中,并补充等温释放介质5ml,反复操作至剩余凝胶量约为总量的10%。相邻时间点的凝胶重量差即为此期间的凝胶溶蚀量,以凝胶累积溶蚀量对时间作图,即得凝胶的经时溶蚀曲线。溶出样品液经0.45μm微孔滤膜过滤,取续滤液照实施例6第1项下色谱条件进样分析,以药物累积释放量对时间作图,得到药物释放曲线,并按下式计算药物累计释放百分率。
其中cn和ci分别为第n个和第i个取样点测得的药物浓度(μg·ml-1);a为凝胶中的药物总量(μg)。
复方紫草微乳凝胶的经时溶蚀曲线和药物释放曲线结果见图6~7。
分别用零级、一级动力学及higuchi方程对凝胶的体外释放曲线进行模型拟合,结果见表2。
表2复方紫草微乳凝胶体外释药的模型拟合
由方程的r值可知,零级动力学方程对复方紫草微乳-原位凝胶的药物释放曲线拟合较好。药物释放行为符合零级动力学方程,凝胶的溶蚀是控制药物释放的主要因素,复方紫草微乳-原位凝胶具有一定的缓释效果。
以上对本发明的优选实施例及原理进行了详细说明,对本领域的普通技术人员而言,依据本发明提供的思想,在具体实施方式上会有改变之处,而这些改变也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!