一种评估多能干细胞质量的方法与流程
1.本发明属于生物信息学技术领域,具体涉及一种评估多能干细胞质量的方法。
背景技术:
2.随着人类多能干细胞,特别是诱导多能干细胞(ipsc)的快速增长,迫切需要一种可以良好的评价现有多能干细胞质量的方法。目前,主流的评价方式是从细胞的形态、多能性验证、核型的检测和遗传稳定性标记检测等方面去单独评估细胞的质量。该方式需要结合多个技术手段,如具备资质的细胞培养员、动物实验(小鼠畸胎瘤实验)、组织切片染色、qpcr技术(荧光定量pcr)等。一套流程评估下来除了漫长的时间等待,还必须耗费大量的人力物力,且基于主流实验去验证细胞质量的方法有较大的主观性和盲目性,不能形成一套准确的科学体系,现已经严重阻碍了多能干细胞的研究和临床的发展。
3.相关技术研究表明采用qpcr的方法可以验证多能干细胞的遗传稳定性。hesc细胞表面的分子标记可以明确细胞的功能。利用基因的荧光反应有效证明多能干细胞的遗传稳定性质量,这可以作为细胞质量的评价标准之一。另外,转录组测序的基因表达量结果与qpcr的结果高度相关,原理也基本一致。
技术实现要素:
4.本发明旨在至少解决上述现有技术中存在的技术问题之一。为此,本发明提出一种评估多能干细胞质量的方法,能够快速评估临床级细胞质量。
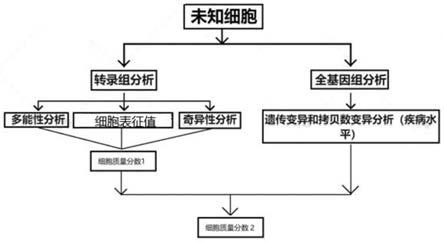
5.根据本发明的一个方面,提出了一种评估多能干细胞质量的方法,所述方法包括以下步骤:
6.s1、对待测细胞进行序列测定,得到转录组表达谱系数据和全基因组序列数据;
7.s2、基于待测细胞的转录组表达谱系数据,进行基因的表达量统计,将基因的表达量统计结果分别进行多能性的分析、奇异性的分析以及与细胞表征值的分析;之后整合细胞多能性的分析、奇异性的分析以及与细胞表征值的分析的结果;得到细胞的质量分数1;基于待测细胞的全基因组序列数据进行全基因组的dna变异分析;
8.s3、根据步骤s2所得的细胞的质量分数和全基因组的dna变异分析结果,得到细胞质量分数2,来评价多能干细胞的细胞质量。
9.在本发明的一些实施方式中,所述多能干细胞包括胚胎干细胞和诱导多能干细胞。
10.在本发明的一些实施方式中,步骤s1中,所述序列测定包括转录组测序,全基因组测序、靶向测序和多重pcr测序、基因芯片和qpcr中的一种。
11.在本发明的一些实施方式中,所述转录组表达谱系数据包括转录组测序数据。
12.在本发明的一些实施方式中,步骤s2中,所述基因表达量的统计还包括对转录组表达谱系数据进行数据分析与质控,将得到的数据与参考基因组进行比对的步骤。
13.在本发明的一些实施方式中,所述数据分析与质控包括使用fastp软件过滤低质
量数据。
14.在本发明的一些实施方式中,所述基因表达量的统计还包括使用fastp软件过滤pair
‑
end的原始低质量rna
‑
seq reads,将pair
‑
end的原始rna
‑
seq reads组装和比对,对齐参考基因组,统计基因表达量,优选地,使用fastp软件过滤低质量数据、hisat2软件组装和比对、使用默认参数对齐参考基因组。
15.在本发明的一些实施方式中,一种评估多组多能干细胞质量的方法,所述方法包括以下步骤:
16.(1)对待测多组细胞进行序列测定,得到转录组表达谱系数据;
17.(2)基于待测细胞的转录组表达谱系数据,进行基因的表达量统计,将基因的表达量统计结果分别进行多能性的分析、奇异性的分析以及与细胞表征值的分析;之后整合细胞多能性的分析、奇异性的分析以及与细胞表征值的分析的结果;得到细胞的质量分数1,对细胞进行排名;
18.(3)选取排名前10%的待测细胞进行序列测定,得到全基因组的序列数据,基于全基因组的序列数据对全基因组的dna变异进行分析;
19.(4)根据步骤(2)和步骤(3)所得的细胞的质量分数和全基因组的dna变异分析结果,得到细胞的质量分数2来评价多能干细胞细胞质量;所述多组为多能干细胞组别大于10。
20.在本发明的一些实施方式中,所述参考基因组为人类参考基因组。
21.在本发明的一些实施方式中,所述参考基因组为人类参考基因组grch37d5。
22.在本发明的一些实施方式中,所述统计基因表达量的使用htseq
‑
count软件和featurecount软件中的一种。
23.在本发明的一些实施方式中,所述统计基因表达量使用htseq
‑
count(v0.13.5)软件的默认参数进行统计(raw count)。
24.在本发明的一些实施方式中,所述多能性分析包括以下步骤:使用r统计语言对多能干细胞和非多能干细胞转录组中的raw count数据进行数据转化,转化方法采用tdm软件,该方法的目的是为了整合不同平台的样本数据,把转化后的rawcount数据变成与微阵列芯片一致的数据,采用lumi(v2.42.0)软件对全部数据进行批次纠正,运用nmf软件对获得的数据矩阵进行nmf(非负矩阵分解)后通过逻辑回归的机器学习模型提取其中的多能性细胞和非多能性细胞数据的特征值,依据训练集和验证集的准确性调整参数,确定分类模型的公式并计算未知细胞的多能性分数。
25.在本发明的一些实施方式中,所述奇异性的分析包括以下步骤:使用r统计语言对esc(胚胎干细胞)转录组中得到的raw count数据进行数据转化,转化方法采用tdm软件,把转化后的raw count数据变成与微阵列芯片一致的数据,该方法的目的是为了整合不同平台的样本数据,采用lumi(v2.42.0)软件对全部数据进行批次纠正,运用nmf软件对获得的数据矩阵进行nmf(非负矩阵分解)后,通过比较分解后的未知样本和胚胎干细胞样本v矩阵的残差和均方根误差(rmse)计算单个得分,作为奇异性评分。
26.在本发明的一些实施方式中,所述细胞表征值的获得包括从转录组表达谱系数据中获取相关细胞遗传稳定基因和细胞功能基因的表达量,使用r统计语言计算与胚胎干细胞样本的基线偏离值。
27.在本发明的一些实施方式中,所述细胞功能基因包括胚胎干细胞表征基因和ips细胞表征基因;所述胚胎干细胞表征基因包括sox2、oct4、nanog、ssea
‑
4、tra
‑1‑
60、tra
‑1‑
81和ssea
‑
1;所述ips细胞表征基因包括ssea3、ssea4、tra
‑1‑
60、tra
‑1‑
81、oct4和nanog。
28.在本发明的一些实施方式中,所述细胞遗传稳定基因包括tert、tet1、tet3、sirt1、chk1、oct4
‑
endo、oct4、nanog和p53。
29.在本发明的一些实施方式中,所述整合细胞多能性的分析、奇异性的分析以及与细胞表征值的分析的结果采用以下公式:
[0030][0031][0032]
c=∑f
i
‑‑‑‑‑‑‑‑‑‑‑
式三;
[0033]
其中,公式一中f
i
是细胞质量评价方面的分数,为评价多能性的分数,x
i
是指某一细胞质量评价下计算得到的值,x
max
是该细胞质量评价设计的最大的阈值,x
min
是该细胞评价设计的最小的阈值,q代表细胞质量评价方面的个数;公式二中f
i
是细胞质量评价方面,为奇异性和细胞表征值中的一种的评价分数,x
i
是指某一细胞质量评价下计算得到的值,x
max
是该细胞质量评价设计的最大的阈值,x
min
是该细胞评价设计的最小的阈值,q代表细胞质量评价方面的个数;公式三中c代表细胞质量分数1。
[0034]
在本发明的一些实施方式中,所述细胞表征值的分析包括以下步骤:从转录组表达谱系数据中获取相关细胞遗传稳定基因和细胞功能基因的表达量,统计计算基因的表达量与胚胎干细胞样本基因的表达量的基线偏离值。
[0035]
在本发明的一些实施方式中,所述统计计算基因的表达量与胚胎干细胞样本的基线偏离值采用r统计语言。
[0036]
在本发明的一些实施方式中,步骤s3中,全基因组数据的处理还包括对全基因组数据进行数据分析与质控步骤,将得到的数据与参考基因组进行比对的步骤。
[0037]
在本发明的一些实施方式中,所述数据分析与质控包括使用fastp软件过滤低质量数据。
[0038]
在本发明的一些实施方式中,全基因组数据处理还包括使用fastp软件处理和清洗pair
‑
end的原始raw reads(fastq)数据,将pair
‑
end的原始raw reads进行组装和比对,对齐参考基因组,对全基因组的变异进行分析,优选地,使用fastp软件处理和分析、bwa软件组装和比对、使用默认参数对齐参考基因组和对全基因组的dna变异进行分析。
[0039]
在本发明的一些实施方式中,所述参考基因组为人类参考基因组。
[0040]
在本发明的一些实施方式中,所述参考基因组为人类参考基因组grch37d5。
[0041]
在本发明的一些实施方式中,步骤s3中,所述全基因组的dna变异分析包括点突变分析、插入缺失分析、拷贝数变异分析、大片段染色体变异分析。
[0042]
在本发明的一些实施方式中,步骤s3中,所述全基因组的dna变异分析采用gatk4软件和cnvnator软件。
[0043]
在本发明的一些实施方式中,所述全基因组的dna变异分析采用gatk4软件的
haplotypecaller组建对经过一系列处理后的bam文件进行snps/indel calling,得到的vcf文件使用专业数据库进行疾病水平的注释。
[0044]
在本发明的一些实施方式中,所述全基因组的dna变异分析采用cnvnator软件对排序后的bam文件进行滑窗分析,得到的vcf文件同样是使用专业的数据库进行疾病水平的注释。
[0045]
在本发明的一些实施方式中,全基因组的dna变异分析包括对全基因组序列数据进行细胞dna突变总数的分析、遗传疾病数目的分析、可能有害变异数目的分析和良性变异数目的分析,之后整合胞dna突变总数的分析、遗传疾病数目的分析、可能有害变异数目的分析和良性变异数目的分析的结果,得到细胞的遗传变异评分。
[0046]
在本发明的一些实施方式中,全基因组的dna变异分析的结果采用以下公式进行计算:
[0047][0048]
v=∑f
i
‑‑‑‑‑‑‑‑‑‑‑‑
式五。
[0049]
其中,公式四中f
i
是细胞变异评价方面的分数,x
i
某一个变异因素下的变异的数目,x
max
为每个变异的因素的最大值,x
min
是每个变异的因素的最小值,公式五中v代表遗传变异评分。
[0050]
在本发明的一些实施方式中,所述变异因素包括突变总数、遗传疾病数目、可能有害变异数目和良性变异数目;x
max
每个变异的因素下各组多能干细胞x
i
中的最小值;x
min
每个变异的因素下各组多能干细胞变异数目x
i
中的最小值。
[0051]
在本发明的一些实施方式中,所述方法还包括对测序前的细胞进行培养步骤,所述步骤包括,将待测细胞使用essential 8tm培养基进行培养。
[0052]
在本发明的一些实施方式中,所述染色体变异包括染色体的缺失、重复、易位、倒位。
[0053]
一种多能干细胞数据库的构建方法,所述方法包括以下步骤:通过整合不同多能干细胞和非多能干细胞的转录组数据构建多能干细胞数据库。
[0054]
一种多能干细胞数据库的构建方法,所述方法包括以下步骤:使用r统计语言对多能干细胞和非多能干细胞转录组中的raw count数据进行数据转化,整合不同测序平台的样本数据,把转化后的raw count数据变成与微阵列芯片一致的数据;采用lumi软件对全部数据进行批次纠正,构建多能干细胞数据库。
[0055]
根据本发明的实施方式,至少具有以下有益效果:本发明方案通过对转录组基因数据和全基因组数据进行生物信息学分析,将所得转录组基因数据和全基因组数据生物信息学分析的结果进行整合,快速计算细胞质量分数;本发明方法简单,且本发明方案是首次全方位的通过细胞的功能和细胞基因组变异的信息对细胞的质量进行全方位数据化地进行评估,能够有效地筛选诱导多能干细胞(ipsc)或者胚胎干细胞(esc)的质量。且本发明采用测序的手段减少了大量实验流程,仅从转录组测序和全基因组测序的层面评估多能干细胞的质量,不仅快速缩短了时间,得到的数据结果通过统计处理可靠准确。
附图说明
[0056]
下面结合附图和实施例对本发明做进一步的说明,其中:
[0057]
图1为本发明实施例1中的转录组表达谱系数据生物信息学分析的流程图;
[0058]
图2为本发明实施例1中的全基因组数据生物信息学分析的流程图;
[0059]
图3为本发明实施例1中的细胞的多能性分析图;
[0060]
图4为本发明实施例1中的评估多能干细胞质量的流程图。
具体实施方式
[0061]
以下将结合实施例对本发明的构思及产生的技术效果进行清楚、完整地描述,以充分地理解本发明的目的、特征和效果。显然,所描述的实施例只是本发明的一部分实施例,而不是全部实施例,基于本发明的实施例,本领域的技术人员在不付出创造性劳动的前提下所获得的其他实施例,均属于本发明保护的范围。
[0062]
细胞培养基:essential 8tm培养基(thermo fisher scientific);测序平台选择mgiseq
‑
2000(购自华大基因)及其配套的全基因组和转录组建库试剂;总rna的提取和dna的提取分别选择试剂盒和purelinktm试剂盒(thermo fisher scientific)。
[0063]
实施例1
[0064]
本实施例提供了一种评估多能干细胞质量的方法,所述方法包括以下步骤:
[0065]
1、将待测细胞采用essential 8tm培养基培养后,采用总rna提取试剂盒(试剂盒)和dna的提取试剂盒(purelink tm试剂盒)进行dna和rna的提取,将提取后的dna和rna进行测序,测序平台选择mgiseq
‑
2000,采用华大基因及其配套的全基因组和转录组建库试剂;得到的转录组表达谱系数据和全基因组序列数据(若是评估多组多能干细胞即采用先提取rna进行测序,将得到的转录组表达谱系数据进行分析,对多组多能干细胞进行排名,选取排名前10%的细胞进行全基因组序列测定,多组即多能干细胞组别大于10组)。
[0066]
2、将步骤1所得的转录组表达谱系数据进行分析:使用fastp软件处理和分析pair
‑
end的原始rna
‑
seq reads(fastq)。对端的reads用hisat2软件进行组装和比对,使用默认参数对齐人类参考基因组(grch37d5)。使用htseq
‑
count(v0.13.5)软件的默认参数进行基因表达量的统计(raw count),转录组表达谱系数据进行生物信息学分析的流程如图1所示。
[0067]
3、将步骤1所得的全基因组数据进行分析:使用fastp软件处理和清洗pair
‑
end的原始raw reads(fastq)数据,使用对端的reads用bwa软件(v0.7.1)进行组装和与参考基因组进行比对,使用默认参数对齐人类参考基因组(grch37d5)形成sam文件。使用samtools(v1.12)软件的对sam文件进行bam文件的转化,方便文件的存储和对其内部的基因组顺序进行排序。全基因组的dna变异分析调用gatk4软件的haplotypecaller组建对经过一系列处理后的bam文件进行snps/indel calling,得到的vcf文件使用专业数据库进行疾病水平的注释。cnv(copy number variation)的流程使用cnvnator软件对排序后的bam文件进行滑窗分析,得到的vcf文件同样是使用专业的数据库进行疾病水平的注释;全基因组数据进行生物信息学分析的流程如图2所示。
[0068]
4、细胞的质量分析
[0069]
(1)多能性的分析
[0070]
使用r统计语言对多能干细胞和非多能干细胞转录组中的raw count数据进行数据转化,转化方法采用tdm软件(thompson,2017),该方法的目的是为了整合不同测序平台的样本数据,把转化后的raw count数据变成与微阵列芯片一致的数据。采用lumi(v2.42.0)软件对全部数据进行批次纠正,运用nmf软件对获得的数据矩阵进行nmf(非负矩阵分解)后通过逻辑回归的机器学习模型提取其中的多能性细胞和非多能性细胞数据的特征值,依据训练集和验证集的准确性调整参数,基于目前的数据集构建逻辑回归模型的多能性最高阈值为94.21,最低阈值为23.344(如图3所示),即高于23.344的细胞均为多能性细胞,低于该阈值甚至到负数为非多能性的细胞,确定分类模型并计算未知细胞的多能性分数,从图中可以看出,本发明方案的分类模型可以准确的区分多能干细胞和非多能干细胞。
[0071]
(2)奇异性的分析
[0072]
使用r统计语言对已知的esc(胚胎干细胞)转录组中得到的raw count数据进行数据转化,转化方法采用tdm软件(thompson,2017),把转化后的raw count数据变成与微阵列芯片一致的数据,该方法的目的是为了整合不同平台的样本数据,采用lumi(v2.42.0)软件对全部数据进行批次纠正,运用nmf软件对获得的数据矩阵进行nmf(非负矩阵分解)后,通过比较分解后的未知样本和胚胎干细胞样本(来自步骤(1)中构建的数据集的高质量胚胎干细胞样本)v矩阵的残差和均方根误差(rmse)计算单个得分,作为奇异性评分。
[0073]
(3)细胞表征值的分析
[0074]
从转录组表达谱系数据中获取相关细胞遗传稳定基因和细胞功能基因的表达量,相关的基因包括胚胎干细胞表征基因(sox2、oct4、nanog、ssea
‑
4、tra
‑1‑
60、tra
‑1‑
81和不表达ssea
‑
1基因),ips细胞表征基因(ssea3、ssea4、tra
‑1‑
60、tra
‑1‑
81、oct4和nanog),细胞遗传稳定基因(ert、tet1、tet3、sirt1、chk1、oct4
‑
endo、oct4、nanog和p53),使用r统计语言计算与胚胎干细胞样本(来自(步骤(1)中构建的数据集中的高质量胚胎干细胞样本))的基因的表达量下的基线偏离值。
[0075]
(4)综合评价可量化的分析
[0076]
将上述多能性、奇异性和细胞表征性分子的偏离值统计成一个细胞质量分数,并进行综合排序,排序公式如下所示:
[0077][0078][0079]
c=∑f
i
‑‑‑‑‑‑‑‑‑‑‑
式三;
[0080]
其中,公式一中f
i
是细胞质量评价方面的分数,为评价多能性的分数,x
i
是指某一细胞质量评价下计算得到的值,x
max
是该细胞质量评价设计的最大的阈值,x
min
是该细胞评价设计的最小的阈值,q代表细胞质量评价方面的个数;公式二中f
i
是细胞质量评价方面,为奇异性和细胞表征中的一种的评价分数,x
i
是指某一细胞质量评价下计算得到的值,x
max
是该细胞质量评价设计的最大的阈值,x
min
是该细胞评价设计的最小的阈值,q代表细胞质
量评价方面的个数(q为3);公式三中c代表细胞质量分数1。
[0081]
多能性分析中最高阈值为94.21,最低阈值为23.344;奇异性分析最高阈值为1.17,最低阈值为0.46;细胞表征值分析中最高阈值为1,最低阈值为0。
[0082]
5、全基因组的dna变异分析(遗传水平的疾病分析)
[0083]
经过细胞质量分数1排名后对需要大规模筛选的细胞挑选排名前10%的细胞进行全基因组测序,目的为了节省测序成本。保证测序的深度大于等于30x,符合市面公认的测序标准。之后对有关cnv和snps/indel的疾病注释结果进行《acmg遗传变异分类标准与指南》分类,标注有遗传风险的细胞,并查阅相关的文献确定真实性。对遗传水平的细胞进行分级评分,确认产生的新的变异并非来自于供体,并且统计该变异的数目,结合《acmg遗传变异分类标准与指南》统计有害的变异、可能良性的变异,以该变异越多细胞越危险作为依据。
[0084][0085]
v=∑f
i
‑‑‑‑‑‑‑‑‑‑‑‑
式五。
[0086]
其中,公式四中f
i
是细胞变异评价方面的分数,x
i
某一个变异因素下的变异的数目,x
max
为每个变异的因素的最大值,x
min
是每个变异的因素的最小值,公式五中v代表遗传变异评分。
[0087]
变异因素包括突变总数、遗传疾病数目、可能有害变异数目和良性变异数目;x
max
每个变异的因素下各组多能干细胞xi中的最小值;x
min
每个变异的因素下各组多能干细胞变异数目xi中的最小值。
[0088]
6、综合评价分析
[0089]
把步骤4得到的值(c)和5得到的值(v)中的结果取平均值作为细胞质量分数2,再次进行排序。
[0090]
本实施例中评估多能性细胞质量的方法的流程如图4所示。
[0091]
测试例1
[0092]
采用上述方法对20个未知多能性的干细胞样本质量进行评估,样本如表1所示。对上述的细胞简称进行转录组,表1中的20组多能干细胞样品培养基均选用essential8
tm
培养基进行培养,测序平台选择mgiseq
‑
2000(华大智造)及其配套的全基因组和转录组建库试剂;总rna的提取和dna的提取分别选择试剂盒和purelink
tm
试剂盒(thermo fisher scientific)。得到转录组表达谱系数据和全基因组序列数据。
[0093]
对c1
‑
c20细胞所得的转录组表达谱系数据按照实施例1中评估多能干细胞质量的方法进行评估干细胞质量。以下是转录组表达谱系数据进行生物信息学分析得到的数据如表2所示。从表中可以看出,不同基因在不同干细胞中的表达量不同。使用r语言统计软件对细胞的多能性、奇异性和细胞表征分子的偏离度进行统计,并统计成细胞质量分数1的评分,并截取前10%的细胞排名,结果如表3所示。
[0094]
对c1
‑
c20细胞所得的全基因组序列数据按照实施例1中评估多能干细胞质量的方法进行评估干细胞质量。全基因组测序数据进行生物信息学分析在疾病水平的注释后并统计变异数据和遗传变异评分如下表4所示。
[0095]
20个未知多能性的干细胞样本质量进行评估如表5所示,细胞质量分数1和遗传变异分数取平均值后,形成细胞质量分数2后再进行排序,可得知这批细胞中最好的是c6。
[0096]
表1
[0097]
样本名细胞类型c1
‑
c60ips细胞
[0098]
表2
[0099]
[0100][0101]
[0102]
[0103][0104]
表3
[0105]
[0106][0107]
表4
[0108]
[0109][0110]
表5
[0111]
[0112][0113]
细胞质量的评估鱼龙混杂,有没有一套科学合理的评估体系。另外,基于主流实验去验证细胞质量的方法有较大的主观性和盲目性,导致花费大量的时间、金钱也可能得不到准确的结果。相关技术提出的多能性和奇异性验证、染色体畸变、细胞遗传稳定性和细胞功能等评估方式都是单一评估模式,没有整合形成一套流程框架。
[0114]
在细胞的多能性和奇异性的评价中,相关技术基于微阵列的基因表达数据集通过机器学习的方式来预测未知细胞样本的多能性。可惜,该方法的弊端也开始日益凸显,现有的测序体系已经开始陆续淘汰原用的微阵列测序技术,取而代之的是新一代的测序技术,使得原有的数据集已经开始无法正确的评估现有的数据。并且,该技术手段受限于当时对胚胎干细胞和ips细胞的理解,培养细胞技术的缺陷,人种和性别之间的差异等问题,该论文并没有进行一定的区分。混杂的实验数据容易发生错误的评估,对于临床级的细胞质量评估是不被允许的。
[0115]
在遗传变异的评估中,相关技术开发的一种可基于转录组表达量数据来评估染色体畸变的方法。然而,转录表达谱仅有全基因组数据的一部分,有关遗传变异的情况并不能从疾病水平上说明问题,从技术层面上来说,对于部分基因组上的微缺失和多缺失,表达谱数据是无法准确呈现,因此会导致一些染色体缺失的情况并不会导致疾病发生的情况出现,也会因为精度的缺失导致疾病的发生却没检测得到,这样会错误评估细胞质量的安全性问题。
[0116]
在细胞表征性分子(细胞遗传稳定和细胞功能)的评估中,基于qpcr技术的实验需要额外去开展,而采用转录组数据中的评估一样能得到有效的数据。
[0117]
单一的评估方式容易忽视其他的同样重要的细胞质量方面,急需一种创新的、可综合以上因素的评估方式。
[0118]
目前,在对细胞进行多能性评价时,小鼠的畸胎瘤试验仍被认为是该领域的金标准,同时也是所有多能干细胞(psc)的核心定义特征,但这一特性的评估方式因没有统一的实验标准受到了科研工作者的质疑。小鼠的畸胎瘤实验主要是在免疫缺陷小鼠的体内注射多能性干细胞后产生分化良好的畸胎瘤。尽管该方法不定量且主观,只有熟练的病理学家
掌握了畸胎瘤组织学可以区分主要由低分化的神经外胚层组成的肿瘤和由所有三个胚胎胚层的高分化组织组成的囊性肿块。前者表现为恶性肿瘤,类似于畸胎瘤,而后者表现为包膜良性肿块,是来自多能干细胞的真正畸胎瘤。由于开展试验的需时长且繁琐,以及对动物的使用和专家的病理评估的要求,畸胎瘤试验作为常规筛查工具有实际的局限性。随着微阵列(芯片)测序技术的发展,有研究采用了一种经济有效的、不含动物的替代畸胎瘤检测方法来评估人类细胞的多能性,该方法可以基于微阵列的基因表达数据集通过机器学习的方式来预测未知细胞样本的多能性。可惜,该方法的弊端也开始日益凸显,现有的测序体系已经开始陆续淘汰原用的微阵列测序技术,取而代之的是二代测序技术或三代测序技术的形式,使得原有的数据集已经开始无法正确的评估现有的数据。并且,受限于当时对胚胎干细胞和ips细胞的理解,培养细胞技术的缺陷,人种和性别之间的差异等问题,若仍旧采用该方法去评估中国人的胚胎干细胞和ips细胞,会出现较大的偏颇,导致不准确的现象时常发生。
[0119]
对于染色体核型的检测,大部分的实验依旧采用组织切片染色技术,该技术能在显微镜下直观的观察染色体的结构和数量。但是,在需要大量筛选临床和科研用途的多能性干细胞时,该方式已然不是最好的选择。相关技术开发了一种可基于转录谱数据来评估染色体畸变的方法,以接近总样本的基因表达量平均值或snps(单核苷酸多态性)的变异程度作为衡量是否发生染色体变异的标准。与全基因组测序下的拷贝数变异和突变分析结果相比较,全基因组测序覆盖的范围更广、分辨精度更高,对于单个样本的结果可以注释到疾病水平上,这是转录组数据中不能比拟的。
[0120]
上面结合附图对本发明实施例作了详细说明,但是本发明不限于上述实施例,在所属技术领域普通技术人员所具备的知识范围内,还可以在不脱离本发明宗旨的前提下作出各种变化。此外,在不冲突的情况下,本发明的实施例及实施例中的特征可以相互组合。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1