一种基于深度学习的蛋白质相互作用位点预测方法
1.本发明涉及生物信息学分析技术领域,具体涉及一种基于深度学习的蛋白质相互作用位点预测方法。
背景技术:
2.作为细胞中最常见的分子之一,蛋白质对于调节细胞中的各种新陈代谢途径以及众多生物学过程具有十分重要的意义。一般来说,蛋白质并不是单独发挥作用的,而是通过彼此之间发生相互作用,即蛋白质
‑
蛋白质相互作用(protein
‑
protein interactions,下文简称ppis)来完成相应的任务。除此以外,对蛋白质相互作用的研究能够为医学诊断和治疗提供新视角,促进新药的设计以及生物医学的发展。因此,预测ppi已成为系统生物学的基础课题,且引起了越来越多的关注。
3.目前,预测蛋白质相互作用的方法主要包括生物学方法和计算学方法两种,在传统的生物学领域,相互作用数据的收集可通过酵母双杂交、蛋白质芯片、合成致死分析等方法完成,然而,这些方法既耗时又费力,导致预测效率不足,且预测结果中经常能观察到该比例的假阴性和假阳性现象。因此,随着计算机技术的高速发展,原本作为辅助手段的计算学方法,目前已经成为预测蛋白质相互作用的主流方法。
4.经调研,已有很多用于预测蛋白质与蛋白质界面相互作用方法被提出,公开号为cn111210871a、公布日为2020年05月29日的中国发明专利申请公开了基于深度森林的蛋白质
‑
蛋白质相互作用预测方法,融合蛋白质对的序列信息、物理化学性质信息和进化信息作为样本的初始特征,使用弹性网进行特征选择,剔除冗余和不相关的特征,将融合后的最优特征向量输入到构建的多粒度级联深度森林中,预测蛋白质
‑
蛋白质相互作用。公开号为cn112259157a、公布日为2021年01月22日的中国发明专利申请公开了一种基于融合生物语义的非相互作用蛋白质对的采样策略的蛋白质相互作用预测方法,基于go术语语义相似度对处于不同分子功能、生物学进程、细胞成分的蛋白质对进行采样并组合获得nips子集。
5.上述方法中,目前仍存在亟待解决的难题,制约着蛋白质相互作用预测的发展:(1)蛋白质的特征提取和序列信息表示;(2)蛋白质相互作用样本数据的不均衡性影响;(3)如何有效地选择和设计ppis分类器;(4)已有预测模型不能很好地满足蛋白质相互作用海量数据的需求。为此,提出一种基于深度学习的蛋白质相互作用位点预测方法。
技术实现要素:
6.本发明所要解决的技术问题在于:如何解决上述背景技术中所提出的问题,提供了一种基于深度学习的蛋白质相互作用位点预测方法,根据蛋白质的一级序列信息提取特征,消除数据集类别不平衡的影响,利用变分自编码器和多层感知机算法对蛋白质残基进行预测。
7.本发明是通过以下技术方案解决上述技术问题的,本发明包括以下步骤:
8.s1:收集数据
9.基于公开的蛋白质相互作用数据集作为基准数据集,获取蛋白质的序列信息;
10.s2:特征提取
11.根据蛋白质序列信息生成位置特异性得分矩阵,提取蛋白质序列的理化特征;
12.s3:特征融合
13.利用滑动窗口技术提取残基与其相邻残基的特征,将提取的所有特征合并成一个特征空间数据集,再结合每一个残基的标签形成模型的输入;
14.s4:均衡处理
15.利用降采样方法采样出具有代表性的特征,得到类别均衡的数据集从而用于训练模型;
16.s5:建立分类器
17.将类别均衡的数据集按比例分为训练集和测试集,将训练集利用变分自编码器进一步提取蛋白质序列的高级抽象特征,利用多层感知机对氨基酸残基进行分类;
18.s6:评估并验证模型
19.将训练好的模型在测试集上测试,得到预测结果;并使用一个公开的独立数据集作为验证集,对模型的鲁棒性进行验证。
20.更进一步地,在所述步骤s1中,所述基准数据集为dset186和dtestset72,分别含有186个和72个蛋白质序列,其中dset186用于训练模型,dtestset72用于作为独立的验证集验证模型,将数据集中标记为相互作用位点的氨基酸残基作为正样本,标记为非相互作用位点的氨基酸残基作为负样本。
21.更进一步地,在所述步骤s2中,所述数据集中的蛋白质序列特征由以下三种组成:位置特异性得分矩阵(pssm)特征、亲水性指数(hi)和相对溶剂可及性(rsa);pssm是通过psi
‑
blast生成的一个20维矩阵,用来描述20种氨基酸的进化保护信息,hi和rsa均为1维数值的蛋白质序列特征。
22.更进一步地,所述步骤s3的具体处理过程:
23.s31:对pssm特征放置一个窗口大小为9的滑动窗口,对于每行残基来说,提取此行残基及其相邻8行残基的特征值,再对这些特征求其平均值作为此行残基的更新值,将滑动窗口依次作用于每行残基,由此得到另一个20维的特征向量,加上之前原始无滑动窗口作用的20维pssm特征,则pssm特征共有40维;
24.s32:分别使用窗口大小为1、3、5、7、9的滑动窗口通过对特征值取平均值的方法得到5维的亲水性指数特征向量和5维的相对溶剂可及性特征向量;
25.s33:将扩充的特征数据组合在一起,每一行特征表示一个氨基酸残基,每个残基具有50维的特征,加上所提取的氨基酸残基的标签,组成一个51维的数据集作为模型的输入。
26.更进一步地,在所述步骤s33中,氨基酸残基的标签值为
‑
1或1,
‑
1代表为负样本,即非相互作用残基;1代表为正样本,即相互作用残基。
27.更进一步地,所述步骤s4的处理过程为:利用nearmiss降采样算法,通过k近邻规则度量正负样本之间的距离,挑选并保留那些到正样本中的最远样本的平均距离最小的负样本,直至正负样本即相互作用残基与非相互作用残基的比例为1:1。
28.更进一步地,在所述步骤s5中,将类别平衡后的数据集按照8:2的比例分为训练集
和测试集,利用训练集训练模型;所述模型包括依次连接的变分自编码器与多层感知机分类器,训练时将训练集送入变分自编码器(vae)中,利用神经网络自动学习数据特征,并且消除数据中的冗余特征,最终提取网络中间隐藏层的30维的抽象特征用于下游的分类任务;将30维的抽象特征再进入多层感知机(mlp)分类器来识别残基是否属于相互作用残基。
29.更进一步地,所述变分自编码器包括编码器与解码器,在所述编码器中,输入数据依次连接一个神经元个数为512的全连接层fc1和一个dropout层,再经过两个全连接层分别得到具有30个高斯分布的均值(z_mean)和方差的对数(z_log_var),引入服从高斯分布的随机噪声(epsilon),利用lambda层将epsilon、z_mean、z_log_var进行线性融合得到中间的隐变量z,这个过程称为采样过程;在所述解码器中,中间的隐变量z经过上述的全连接层fc1和一个dropout层,再连接一个全连接层输出一个近似于输入数据的50维数据。
30.更进一步地,所述多层感知机分类器包括一个lambda层、三个全连接层和一个dropout层,lambda层主要将均值(z_mean)和方差的对数(z_log_var)结合起来用于神经网络数据的传递,最后利用softmax函数对残基进行二分类。
31.更进一步地,在所述步骤s6中,将训练好的模型在测试集上进行测试,得到模型预测的准确率、召回率、精确率、f1
‑
值、mcc值;并利用dtestset72作为独立验证集,通过s1~s6同样的步骤且不改变模型的任何参数来处理dtestset72数据集,得到模型的评估指标,以此验证模型的泛化能力。
32.本发明相比现有技术具有以下优点:该一种基于深度学习的蛋白质相互作用位点预测方法,基于nearmiss降采样算法对样本数据分布不均衡问题进行处理,从而可以避免模型为了最大限度地提高预测准确度而偏向于多数类,忽略少数类
‑
即相互作用残基的预测准确度,nearmiss降采样算法是一种根据度量样本之间的距离删除部分多数类的抽样方法,它的抽样过程考虑到了全局信息,从而使得被留下的多数类数据更具代表性,另一方面,这种降采样方法也会提高模型的运行速度;使用变分自编码器器对特征空间数据集进一步的提取和压缩,变分自编码器是一种无监督的学习算法,将输入信息进行压缩,提取出数据中最具代表性的信息,其目的是在保证重要特征不丢失的情况下,降低输入信息的维度,减小神经网络的处理负担,简单来说就是提取输入信息的更高级更抽象的特征,方便于后续分类工作的进行,值得被推广使用。
附图说明
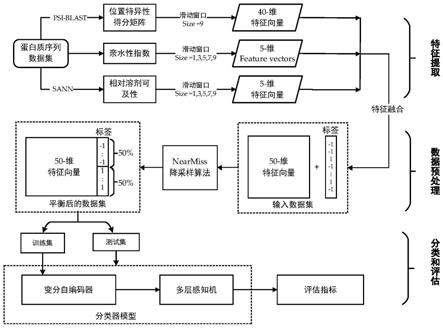
33.图1是本发明实施例中一种基于深度学习的蛋白质相互作用位点预测方法的流程示意图;
34.图2是本发明实施例中变分自编码器与多层感知机分类器相配合示意图。
具体实施方式
35.下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
36.本实施例提供一种技术方案:一种基于深度学习预测蛋白质相互作用位点的方法,根据蛋白质的一级序列信息提取特征,消除数据集类别不平衡的影响,利用变分自编码
器和多层感知机算法对蛋白质残基进行预测。
37.结合图1所示,本发明的一种基于深度学习预测蛋白质相互作用位点的方法,具体步骤如下:
38.1)下载公开的蛋白质相互作用数据集作为基准数据集,获取蛋白质的序列信息。
39.本实施例中选用的数据集来源于公开的基准数据集dset186和dtestset72,这两个数据集是由日本研究者murakami和mizuguchi于2010年开发出来的,它们分别含有186个和72个蛋白质序列,其中dset186被用作训练模型,dtestset72作为独立的验证集验证模型。对于蛋白质氨基酸来说,若其与其他氨基酸结合后失去的绝对溶剂可及性面积小于则表示此氨基酸为相互作用残基,否则,为非相互作用残基。将数据集中标记为相互作用位点的氨基酸残基作为正样本,记为1;标记为非相互作用位点的氨基酸残基作为负样本,记为
‑
1。dset186和dtestset72中的相互作用残基数量与非相互作用残基数量及相互作用残基占总残基数量的比例如表1所示。
40.表1数据集中的残基数量
[0041][0042]
2)根据蛋白质序列信息生成位置特异性得分矩阵,提取蛋白质序列的理化特征。
[0043]
数据集中的蛋白质序列特征由以下3种组成:位置特异性得分矩阵(简称pssm)特征,亲水性指数(简称hi)和相对溶剂可及性(简称rsa)。pssm是通过运行psi
‑
blast算法在ncbi的非冗余(nr)序列数据库中搜索生成的,有三个迭代,e值阈值为0.001。每个氨基酸被编码为一个含有20个元素的载体。pssm或特定于位置的评分矩阵是蛋白质blast搜索中使用的一种评分矩阵,其中蛋白质多序列比对中每个位置的氨基酸取代分数分别给出。因此,比对中位置a处的tyr
‑
trp取代与位置b中相同的取代可能获得非常不同的分数,pssm分数通常显示为正整数或负整数。正值表示给定的氨基酸替换比对发生的频率比偶然预期的要高,而负值表示替换发生的频率低于预期的发生率。通过输入蛋白质序列的fasta文件可查看pssm。亲水性指数是ppis鉴定的另一个重要特征,某种氨基酸的亲水指数是一个描述其支链的亲水性或疏水性程度大小的值。“亲水指数”于1982年被jack kyte与russell doolittle提出,亲水指数越大,这种氨基酸的疏水性就越强。由于蛋白质溶剂可及表面积或大或小,简单地通过其数值的大小不能准确衡量其物理性质,所以将溶剂可及表面积转化为溶剂可及性进一步分析蛋白质的结构和性质。相对溶剂可及性是通过使用在线服务器sann测定的。hi和rsa均为1维数值的蛋白质序列特征。
[0044]
3)考虑到蛋白质链中氨基酸残基是否为相互作用残基与其相邻的残基的性质密切相关,利用滑动窗口技术提取残基与其相邻残基的特征,将提取的所有特征合并成一个特征空间数据集,再结合每一个残基的标签形成模型的输入。
[0045]
滑动窗口大小为(2n+1)表示我们考虑位于中心的目标氨基酸和2n个相邻的氨基酸作为目标氨基酸的输入特征。首先对pssm特征放置一个窗口大小为9的滑动窗口,对于每行残基来说,提取此行残基及其相邻8行残基的特征值,再对这些特征求其平均值作为此行残基的更新值,将滑动窗口依次作用于每行残基,由此得到另一个20维的特征向量,加上之
前原始无滑动窗口作用的20维pssm特征,则pssm特征共有40维。然后,分别使用窗口大小为1、3、5、7、9的滑动窗口通过对特征值取平均值的方法得到5维的亲水性指数特征向量和5维的相对溶剂可及性特征向量。对于一个给定的蛋白质序列,经过滑动窗口后蛋白质链的首尾四个残基将会被丢弃,因为位于首和尾的4个残基无法使用窗口大小为9的滑动窗口。将扩充的特征数据组合在一起,每一行特征表示一个氨基酸残基,每个残基具有50维的特征,加上所提取的氨基酸残基的标签,组成一个51维的数据集作为模型的输入。氨基酸残基的标签值为
‑
1或1,其中
‑
1代表为负样本,即非相互作用残基;1代表为正样本,即相互作用残基。
[0046]
4)为了解决数据集的类别不平衡问题,利用降采样技术采样出具有代表性的特征,得到类别均衡的数据集从而用于训练模型。
[0047]
使用样本分布不均衡的数据集将会导致模型的准确性和鲁棒性很差,这是ppis预测中比较常见的问题。对数据集的类别不平衡性的处理为:利用nearmiss降采样算法,通过k近邻规则度量正负样本之间的距离,nearmiss算法根据规则的不同可分为三种版本,实验结果表明当版本设为2的时候预测效果最佳,版本2的规则是挑选并保留那些到正样本中的最远样本的平均距离最小的负样本,直至正负样本即相互作用残基与非相互作用残基的比例为1:1。
[0048]
5)将类别均衡的数据集按比例分为训练集和测试集,将训练集利用变分自编码器进一步提取蛋白质序列的高级抽象特征,再利用多层感知机对氨基酸残基进行分类。
[0049]
首先将类别平衡后的数据集按照8:2的比例分为训练集和测试集,利用训练集训练模型。将训练集送入构建的变分自编码器(简称vae)中,利用神经网络自动学习数据特征,并且消除数据中的冗余特征,最终提取网络中间隐藏层的30维的抽象特征用于下游的分类任务。由于变分自编码器提取的特征已经非常具有代表性,所以在变分自编码器后接一个简单的多层感知机(简称mlp)分类器来识别残基是否属于相互作用残基。
[0050]
结合图2所示,vae的构建方法为:设计一个无监督学习网络,其输入和输出均为蛋白质数据集分割后的训练集,通过深度神经网络尽量使得输入和输出的相似度高。本发明是在变分自编码器网络上添加了一个softmax多层感知器,以同时获得降维和分类输出。首先将训练集的50维特征数据通过编码器得到中间隐藏层,再通过解码器重构出输入的50维特征向量。所述变分自编码器包括编码器与解码器,在所述编码器中,输入数据依次连接一个神经元个数为512的全连接层fc1和一个dropout层,再经过两个全连接层分别得到具有30个高斯分布的均值(z_mean)和方差的对数(z_log_var),引入服从高斯分布的随机噪声(epsilon),利用lambda层将epsilon、z_mean、z_log_var进行线性融合得到中间的隐变量z,这个过程称为采样过程;在所述解码器中,中间的隐变量z经过上述的全连接层fc1和一个dropout层,再连接一个全连接层输出一个近似于输入数据的50维数据。设计神经网络中间隐藏层的维数为30,低于原先构建的数据集维数50维,这样既提取到了数据中有用的特征又降低了特征的维数,使得模型的速度更快且预测准确率更高。vae使用kl散度作为损失函数计算重构误差。
[0051]
多层感知机分类器包括一个lambda层、三个全连接层和一个dropout层,lambda层主要将均值(z_mean)和方差的对数(z_log_var)结合起来用于神经网络数据的传递,最后利用softmax函数对残基进行二分类。
[0052]
6)将训练好的模型在测试集上测试,得到模型预测准确率、召回率、f1
‑
值等评估指标。为了验证模型的有效性,再使用一个公开的独立数据集作为验证集,对模型的鲁棒性进行验证。
[0053]
通过上述分类模型即可得到蛋白质相互作用位点,进一步地,利用测试集对得到的蛋白质相互作用位点进行测试评价。其中,测试的指标为:
[0054]
准确率:
[0055][0056]
召回率:
[0057][0058]
精确率:
[0059][0060]
f1
‑
值:
[0061][0062]
mcc值:
[0063][0064]
其中tp为真阳性数目,为正确预测出来的正样本数目;tn为真阴性数目,表示正确预测出来的负样本数目;fp为假阳性数目,即预测结果中,本来是负样本被预测为正样本的数目;fn为假阴性数目,即本来是正样本而被错误预测为负样本的数目。f1
‑
值是precision和recall的加权调和平均,即综合了precision和recall的结果,当f1值较高时说明试验方法比较有效;mcc是衡量不平衡问题的很好地衡量标准,本质是真实值和预测值之间的相关系数,在
‑
1和1之间,
‑
1表示预测结果最差,1表示预测结果最好。
[0065]
本实施例对蛋白质相互作用位点识别的结果详见表2所示。
[0066]
表2模型的分类性能评估
[0067]
数据集准确率召回率精确率f1
‑
值mccdset1860.8550.7580.9380.8380.722dtestset720.7630.6800.8230.7440.535
[0068]
由表2中数据可以看出本实施例的dset186的分类准确率达到了85.5%,召回率达到了75.8%,说明相互作用残基被正确预测的效果达到较好的水平。f1
‑
值和mcc分别达到83.8%和72.2%,说明模型总体分类性能较好,能够较准确的预测数残基是否为相互作用残基或非相互作用残基。由于dtestset72的数据较少,所以各评估指标不如dset186,但也处于较好的水平,所以模型具有较好的鲁棒性。
[0069]
为了进一步评价此模型的性能,将其与psiver、loris、crf、sswrf这4种现有模型进行了比较。将此模型记为vaemlp。同样在dset186和dtestset72数据集上,vaemlp与其他
模型的评估指标比较如表3所示。
[0070]
表3与现有蛋白质相互作用预测方法的比较
[0071][0072]
表3中的结果充分说明,本发明构建的模型各类评价指标得到显著提高,能够有效识别蛋白质相互作用位点。
[0073]
综上所述,上述实施例的一种基于深度学习的蛋白质相互作用位点预测方法,基于nearmiss降采样算法对样本数据分布不均衡问题进行处理,从而可以避免模型为了最大限度地提高预测准确度而偏向于多数类,忽略少数类
‑
即相互作用残基的预测准确度,nearmiss降采样算法是一种根据度量样本之间的距离删除部分多数类的抽样方法,它的抽样过程考虑到了全局信息,从而使得被留下的多数类数据更具代表性,另一方面,这种降采样方法也会提高模型的运行速度;使用变分自编码器器对特征空间数据集进一步的提取和压缩,变分自编码器是一种无监督的学习算法,将输入信息进行压缩,提取出数据中最具代表性的信息,其目的是在保证重要特征不丢失的情况下,降低输入信息的维度,减小神经网络的处理负担;简单来说就是提取输入信息的更高级更抽象的特征,方便于后续分类工作的进行。
[0074]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1