一种基于可变聚腺苷酸化位点的疾病预后标志物筛选方法
1.本发明涉及高通量测序、基因可变聚腺苷酸化位点识别和疾病预后风险基因筛选技术领域。具体涉及一种基于可变聚腺苷酸化表达数据的预后风险基因组筛选方法及其应用。
背景技术:
2.可变聚腺苷酸化(alternative poyadenylation,apa)是基因的一种主要转录后调节方式。apa位点常发生于基因的3’非编码区域,可产生不同3’非编码区末端的转录本,在poly(a)聚合酶的作用下生成不同位置和长度的poly(a)尾,影响rna加工因子和rna结合蛋白等调节分子与转录本的结合,进而影响mrna的稳定性及不同转录本的表达。已有研究发现,apa具有显著的组织特异性,在细胞的增殖和分化中具有重要作用。
3.恶性肿瘤作为一种与细胞的异常分化和增殖密切相关的疾病,其细胞中一些基因的apa位置及转录本表达量与正常细胞存在显著差异。例如,在8号染色体和21号染色体易位白血病(t(8;21)aml)的细胞中,近端apa导致aml1
‑
ae融合基因3’utr区域的缩短并提高ae基因的稳定性,促进白血病细胞的增长,影响患者的治疗和预后。这提示apa差异基因具有作为肿瘤治疗靶点和患者预后预测指标的潜在可能性。在临床应用中,患者的预后分层在用药选择、疗效评估、复发监测等方面具有重要指导作用。
4.随着高通量测序技术的发展,全转录组测序越来越多的应用于复杂疾病的临床诊断和治疗中,产生的大量测序数据也为深入挖掘疾病生物机制提供了基础。由于组学数据具有小样本、高维度的特征,为了获得更加稳定、可解释的模型,从大量数据中筛选与问题密切相关的特征子集,是生物信息数据挖掘中的重要步骤。
5.逐步回归是一种常用的变量选择方法,其将变量逐个引入模型或逐个从模型中去除,基于赤池信息准则(akaike information criterion,aic)等模型评价准则比较引入或删除变量前后的模型性能,并保留使模型性能提高的变量,重复该过程直到不能再引入有效的新变量,得到与预测结果显著相关的变量集。逐步回归适用于特征较多的数据,搜索方法包括前进法、后退法和混合法。其中,前进法和后退法通常只能达到局部最优,混合法可能引入贡献较小的特征,存在过拟合问题。
6.近年来,许多惩罚回归模型也被提出以解决变量回归模型中的不稳定性、计算困难性等问题,这些模型将无关变量的系数收缩至零,将系数非零的变量作为筛选的子集。最常使用的惩罚回归模型是lasso,其将l1惩罚和线性模型结合使部分子集权重为0,相应特征被忽略。相比于lasso回归模型,breheny和huang提出的基于非凸的平滑削边绝对偏离(smoothly clipped absolute deviation,scad)惩罚和最小最大凹惩罚(mimimax concave penalty,mcp)方法,构建了更加稳定的变量选择回归模型,并提供了实现算法的r语言ncvreg包。
技术实现要素:
7.本发明的目的在于提供一种基于可变聚腺苷酸化位点的疾病预后标志物筛选方法,从转录组测序数据中识别转录后调节相关的apa特征,筛选预后相关标志基因组,提供预后风险得分计算公式,基于数据辅助预测临床疾病预后。
8.为了达到上述目的,本发明采用以下技术方案予以实现:
9.步骤一,从样本全转录组测序数据中识别3’非翻译区可apa位点,计算不同转录本的表达量,并过滤表达量过低的apa位点;
10.步骤二,对步骤一所述的apa位点,通过单因素cox回归分析初步筛选可能与预后相关的apa位点,将同一基因上所有apa位点的表达量相加作为该基因的表达量;
11.步骤三,对步骤二所述的基因,基于逐步回归和最小最大凹惩罚的多因素cox回归模型进行进一步的筛选,得到预后风险基因组及相应系数,得到预后风险得分计算公式;
12.步骤四,根据步骤三得到的预后风险得分计算公式预测样本预后为高危组或低危组。
13.优选地,步骤一中,apa位点识别及表达量计算使用apa定量算法(quantification of apa,qapa)。
14.优选地,步骤二中,单因素cox回归分析初步筛选设置纳入阈值为p值<0.01。
15.优选地,步骤三中,逐步回归使用r语言mass包的stepaic函数,搜索方法使用混合法(direction=“both”),基于最小最大凹惩罚的cox回归模型使用r语言ncvreg包的cv.ncvsurv函数(penatly=“mcp”)。模型评价使用aic指标。
16.优选地,步骤四中,使用r语言中plotroc包绘制roc曲线,根据roc曲线确定预后分层最佳阈值,将预后风险得分>最佳阈值设定为预后高危组,将预后风险的风≤最佳阈值设定为预后低危组,使用r语言survminer包中的ggsurvplot函数绘制kaplan
‑
meier曲线比较两组间的生存差异。roc曲线使用r语言中plotroc包的ggplot函数实现;生存分析使用r语言中的survival包实现。
17.通过高通量测序获得新纳入样本的筛选基因转录本表达量,计算预后风险得分,根据分类阈值预测样本属于预后高风险组或预后低风险组。
18.与现有技术相比,本发明具有以下有益效果:
19.目前仅基于高维度、小样本的传统基因表达数据,对复杂疾病的基因表达调控等生物机制的研究程度有限。本发明基于基因可变聚腺苷酸化表达数据,将基因的转录后调控情况作为标志与疾病发展相关联,为疾病的预后进行分层,为复杂疾病的临床干预提供指导信息。本发明基于大量已有临床数据构建稳定回归模型,可对新纳入样本选择标志基因进行测序,避免全转录组测序的高成本、高噪声、复杂分析等问题,便于临床的推广和应用。
附图说明
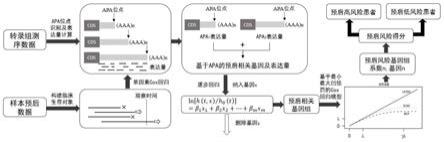
20.图1是基于基因可变聚腺苷酸化表达数据的预后风险基因组筛选方法的流程;
21.图2是根据预后风险得分对样本生存分析的roc曲线;
22.图3是根据预后风险得分的预后分层kaplan
‑
meier曲线。
具体实施方式
23.为了使本技术领域的人员更好地理解本发明方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分的实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
24.需要说明的是,本发明的说明书和权利要求书及上述附图中的术语“第一”、“第二”等是用于区别类似的对象,而不必用于描述特定的顺序或先后次序。应该理解这样使用的数据在适当情况下可以互换,以便这里描述的本发明的实施例能够以除了在这里图示或描述的那些以外的顺序实施。此外,术语“包括”和“具有”以及他们的任何变形,意图在于覆盖不排他的包含,例如,包含了一系列步骤或单元的过程、方法、系统、产品或设备不必限于清楚地列出的那些步骤或单元,而是可包括没有清楚地列出的或对于这些过程、方法、产品或设备固有的其它步骤或单元。
25.下面结合附图对本发明做进一步详细描述:
26.图1是基于基因可变聚腺苷酸化表达数据的预后风险基因组筛选方法的流程,主要包括基因可变据腺苷酸化位点的示意及表达数据的计算示意、单因素cox回归进行预后相关基因的初步筛选、逐步回归和基于mcp的cox回归的进一步基因筛选和疾病预后分层。
27.图2是根据预后风险得分的生存分析受试者工作特征曲线(receiver operating characteristic curve,roc),其横坐标为假阳性率,纵坐标为真阳性率,roc曲线下面积(area under roc curve)越接近1表示模型分类性能越好。根据roc曲线可获得最佳分类阈值。
28.图3是根据预后风险得分的预后分层kaplan
‑
meier曲线,较平缓的曲线说明该组患者生存时间长,预后风险低,较陡峭的曲线说明该组患者生存时间短,预后风险高。
29.本实施例以中国三阴性乳腺癌人群病理组织rna测序数据为样本,进行以下实验。
30.1、预后相关apa位点初步筛选
31.对352例中国三阴性乳腺癌患者转录组测序数据通过apa定量算法(quantification of apa,qapa)识别并注释基因3’非翻译区apa位点位置。对具有多个apa位点的基因,qapa计算每个apa位点对应转录本的每百万碱基读段覆盖(transcripts per million,tpm)作为apa表达量。对于每个转录组测序样本,共注释34074个apa位点。对apa表达量进行对数变换,令,apa表达量=log2(tpm+1)。删除在所有样本中apa表达量平均值低于1的apa位点,剩余apa位点共20736个,位于12858个基因上。
32.将352个样本的生存时间和随访状态与基因可变聚腺苷酸化表达数据合并,使用cox单因素回归分析对三阴性乳腺癌预后相关apa位点进行初步筛选,以p<0.005为纳入标准。将同一基因上apa位点的表达量求和计算基因的总表达量,得到初步筛选的预后风险相关基因共74个。
33.2、筛选预后风险基因组
34.筛选预后风险基因组包括逐步回归模型的构建和基于mcp的cox回归模型构建两部分。使用步骤1中初步筛选获得的74个预后相关基因构建预后风险基因组筛选模块。在逐步回归模型的构建中,首先使用患者的预后信息和基因表达数据构建基于逐步回归的模
型,使用混合法进行变量选择。在混合法中,模型从没有变量开始,使用前进法的方式添加提高模型性能的基因作为变量,使用后退法的方式删除不改善模型性能的基因变量。通过逐步回归模型,共保留预后相关基因49个。
35.使用逐步回归模型筛选获得的49个基因构建基于mcp的cox回归模型。在cox回归模型中,以对患者随访记录的生存时间和状态数据构建生存对象,以49个基因的表达量作为协变量。基于mcp的算法将部分基因的系数收缩到0,筛选得到与三阴性乳腺癌预后风险相关的标志基因集,其中包含基因13个,分别为abt1,aimp1,atp7b,cd55,fam98b,mllt10,nhs,nudt16,pla2g16,pou3f3,ppp2r5e,slc16a11和znf134。预后风险得分(prognosis risk score,prs)计算公式为:
36.prs=
‑
0.783*abt1+1.700*aimp1+0.789*atp7b+0.161**cd55+1.538*fam98b+0.558*mllt10+0.843*nhs
‑
0.987*nudt16+0.517*pla2g16
‑
0.195*pou3f3+0.373*ppp2r5e+0.424*slc16a11
‑
1.223**znf134
37.其中,abt1,aimp1,atp7b,cd55,fam98b,mllt10,nhs,nudt16,pla2g16,pou3f3,ppp2r5e,slc16a11,znf134均表示对应基因基于可变聚腺苷酸化的总表达量。上述预后风险基因组及包含apa事件如表1所示。在筛选获得预后风险相关基因组中,基因abt1和znf134与基因的转录、激活过程相关,fam98b,mllt10和pou3f3被报道分别与大肠癌、白血病、食道癌的发生发展相关,aimp1,pla2g16和ppp2r5e参与ras、ret等重要信号转导通路,参与细胞凋亡、生长、分裂过程的调控。
38.表1
[0039][0040]
3、预后分层及生存分析
[0041]
根据步骤2中的预后风险得分公式对根据基因表达量对每个样本计算相应的预后风险得分prs,根据roc曲线(图2)确定最佳分组阈值,按照最佳分组阈值将样本划分为预后高风险组和预后低风险组。按照预后分层和患者随访时间绘制kaplan
‑
meier曲线如图3所示。按照prs指标划分的预后高风险组和预后低风险组生存时间存在统计学差异(p值<0.0001)。
[0042]
由上述实验可知,本发明能够筛选稳定的复杂疾病预后风险预测基因集,具有可重复性,并具有较好的临床应用性能,便于进行针对少量目标基因的测序及分析,可以降低临床检测成本,辅助临床患者预后的预测。本发明使用不同数据集可构建针对不同疾病的预后分层模型,具有可扩展性。
[0043]
以上实施例仅用于解释说明本发明的技术方案,而非限制本发明的保护范围,凡是按照本发明提出的技术思想,在技术方案的基础上做的修改或同等替换,均落入本发明权利要求书的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1