一种稻瘟菌侵染水稻关键sRNA识别系统及识别方法
一种稻瘟菌侵染水稻关键srna识别系统及识别方法
技术领域
1.本发明属于生物信息学技术领域,特别涉及一种稻瘟菌侵染水稻关键srna识别系统及识别方法。
背景技术:
2.由稻瘟菌引起的稻瘟病对全世界的稻米及其它禾谷类作物生产构成巨大危害。尽管稻瘟菌是研究植物真菌病害的模式真菌,然而目前研究表明,在田间使用水稻杀真菌剂或选用抗病水稻品种对稻瘟病害的长期控制的表现中仍不稳定。为了找到对稻瘟病持久有效的防治方法,关键在于揭示稻瘟菌侵染水稻的细胞机制。
3.随着真菌
‑
植物互作相关的转录组学、基因组学、蛋白质组学和代谢组学等组学数据大量出现,研究者开始基于组学数据,应用生物计算方法辅助和指导生物实验揭示生物分子间相互作用,挖掘生物过程中的关键因子。目前多组学数据整合方法的应用在癌症等复杂疾病的预后分析、分类等方面有一定成效,但在真菌
‑
植物互作机制的研究还处于起步阶段。
4.如今,深度学习在各大领域的预测问题上均有非常有效的结果。深度学习的方法可以在大规模数据中,通过构建多组学分层异质网络来提取出有效的、隐含的特征,并利用这些特征构建出有效的预测模型。目前,深度学习的方法在生物信息学有了较大的突破。因此,将深度学习方法应用到基于多组学数据识别关键致病srna成为一个新的研究领域。
5.目前,识别稻瘟菌关键srna的方法存在如下问题:
6.(1)尽管稻瘟菌与水稻组学数据日益丰富,但如何整合各组学数据平台资源并加以分析仍是难题;
7.(2)病原菌srna如何参与到真菌
‑
植物互作过程,与各组学致病标志物之间的调控关系尚不为人所知。
技术实现要素:
8.本发明的目的之一是提供一种稻瘟菌侵染水稻关键srna识别系统,其能够准确识别稻瘟菌、水稻多组学数据,并识别稻瘟菌致病相关的关键srna。
9.本发明还提供了一种稻瘟菌侵染水稻关键srna识别方法,其联合致病因子关联网络从统计角度对跨物种的关键srna进行挖掘,能够对稻瘟菌关键致病srna进行更准确的识别。
10.本发明提供的技术方案为:
11.一种稻瘟菌侵染水稻关键srna识别系统及识别方法,包括:
12.输入单元,其用于输入稻瘟菌和水稻多组学数据;
13.微处理器,其连接所述输入单元;
14.存储单元,其连接所述微处理器;
15.处理单元,其连接所述微处理器,用于处理数据并得出识别结果;
16.其中,所述处理单元包括:
17.预处理单元,其从所述存储单元中获取所述多组学数据,并进行预处理;
18.网络构建单元,其获取预处理后的多组学数据,处理得到稻瘟菌和水稻多组学分层异质互作网络;
19.致病因子挖掘单元,其输入所述多组学分层异质互作网络,输出致病srna调控模块;以及
20.关键srna识别单元,其接收致病srna调控模块,并进行稻瘟菌关键srna识别,得到识别结果。
21.优选的是,所述的稻瘟菌侵染水稻关键srna识别系统,还包括:
22.第一接口单元,其连接所述输入单元,所述第一接口单元包括第一usb接口、第一jtag调试接口、第一以太网接口和第一rs
‑
232接口;以及
23.第二接口单元,其连接所述预处理器,所述第二接口单元包括第二usb接口、第二jtag调试接口,第二以太网接口和第二rs
‑
232接口;
24.其中,所述第二jtag调试接口与所述第一jtag调试接口相连,所述第二以太网接口与所述第一以太网接口相连,所述第二rs
‑
232接口与所述第一rs
‑
232接口相连。
25.一种稻瘟菌侵染水稻关键srna识别方法,包括如下步骤:
26.步骤一、采集稻瘟菌和水稻多组学数据;
27.其中,所述多组学数据包括:基因组学数据、转录组学数据、蛋白组学数据和代谢组学数据;
28.步骤二、利用所述多组学数据构建稻瘟菌和水稻多组学分层异质互作网络;
29.步骤三、对所述稻瘟菌和水稻多组学分层异质互作网络进行稻瘟菌srna与水稻mrna差异表达数据分析,得到稻瘟菌侵染水稻过程中的跨物种致病srna调控模块;
30.步骤四、通过所述致病srna调控模块识别稻瘟菌致病相关的关键srna。
31.优选的是,在所述步骤二中,构建稻瘟菌和水稻多组学分层异质互作网络,包括如下步骤:
32.步骤1、在基因组学中,建立稻瘟菌基因和水稻基因互作调控网络;在转录组学中建立稻瘟菌srna和水稻mrna互作网络;在蛋白组学中,建立稻瘟菌蛋白和水稻蛋白组数据互作网络;在代谢组学中,建立水稻和稻瘟菌代谢组学关系网络;
33.步骤2、采用基于多组学数据的聚类降维算法将所述稻瘟菌基因和水稻基因互作调控网络、所述稻瘟菌srna和水稻mrna互作网络、所述稻瘟菌蛋白和水稻蛋白组数据互作网络、所述水稻和稻瘟菌代谢组学关系网络进行纵向整合,建立初级稻瘟菌
‑
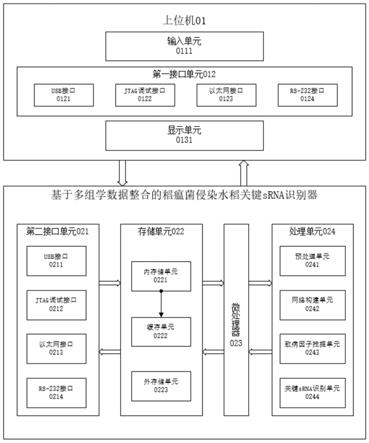
水稻分层异质互作网络;
34.步骤3、对所述稻瘟菌基因和水稻基因互作调控网络、所述稻瘟菌srna和水稻mrna互作网络、所述稻瘟菌蛋白和水稻蛋白组数据互作网络、所述水稻和稻瘟菌代谢组学关系网络进行纵向整合,建立初级稻瘟菌
‑
水稻分层异质互作网络分别通过上下游组学数据库进行扩充,获取相应的调控网络作为演示网络;对所述初级稻瘟菌
‑
水稻分层异质互作网络进行优化,完善稻瘟菌
‑
水稻互作知识数据库,得到稻瘟菌
‑
水稻多组学分层异质互作网络。
35.优选的是,在所述步骤1中,建立稻瘟菌蛋白和水稻蛋白组数据互作网络,包括:
36.根据所述蛋白组学数据得到差异表达的蛋白质,将差异表达的蛋白质输入到
string数据库,建立水稻ppi网络构建和稻瘟菌ppi网络构建;
37.使用基于图的支持向量机方法完成节点选取和网络可视化并为节点赋予标签属性;
38.将网络中的正标签节点进行筛选并可视化蛋白质组学数据网络,得到稻瘟菌
‑
水稻蛋白组数据互作网络。
39.优选的是,在所述步骤三中,通过建立稻瘟菌侵染水稻过程中的跨物种致病srna调控网络,得到稻瘟菌侵染水稻过程中的跨物种致病srna调控模块;
40.其中,建立稻瘟菌侵染水稻过程中的跨物种致病srna调控网络,包括如下步骤:
41.步骤a、采用op
‑
cluster算法构建初级致病调控网络;
42.步骤b、在多组学分层异质网络中获得整合数据,筛选致病相关的多组学数据输入到inmf多组学联合分析中,对所述初级致病调控网络进行修剪,得到稻瘟菌侵染水稻过程中的跨物种致病srna调控网络。
43.本发明的有益效果是:
44.(1)本发明提供的稻瘟菌侵染水稻关键srna识别系统,能够准确识别稻瘟菌、水稻多组学数据,并识别稻瘟菌致病相关的关键srna。
45.(2)本发明提供的稻瘟菌侵染水稻关键srna识别方法,联合致病因子关联网络从统计角度对跨物种的关键srna进行挖掘,能够对稻瘟菌关键致病srna进行更准确的识别。
附图说明
46.图1本发明所述的稻瘟菌侵染水稻关键srna识别系统的结构示意图。
47.图2本发明所述的稻瘟菌侵染水稻关键srna识别器的模型原理图。
48.图3本发明所述的稻瘟菌侵染水稻关键srna识别器的电路原理图。
49.图4本发明所述的稻瘟菌侵染水稻关键srna识别方法的流程图。
50.图5本发明所述的多组学分层异质网络横向构建流程图。
51.图6本发明所述的局部双向聚类算法op
‑
cluster与全局多组学联合分析算法inmf流程图。
具体实施方式
52.下面结合附图对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
53.如图1所示,本发明提供了一种基于多组学数据整合的稻瘟菌侵染水稻关键srna识别系统,上位机01由输入单元0111,第一接口单元012的usb接口0121、jtag调试接口0122、以太网接口0123、rs
‑
232串口0124,显示单元0131共同构成来完成与arm9微处理器023的协调工作。其中,输入单元0111与第一接口单元012连接,负责完成稻瘟菌和水稻多组学数据的输入;第一接口单元012负责与arm9微处理器023进行连接通信;显示单元0131与第一接口单元012连接,负责完成稻瘟菌关键srna识别结果的输出显示。
54.稻瘟菌侵染水稻关键srna识别器由arm9微处理器023、第二接口单元021、存储单元022以及处理单元024构成;其中,第二接口单元021包括usb接口0211、jtag调试接口0212、以太网接口0213及rs
‑
232串口0214;usb接口0211可与u盘连接,实现将稻瘟菌关键
srna识别得到的结果数据的转存,以此实现存储单元的扩增;jtag调试接口0212通过jtag仿真(编程器)转换设备与上位机jtag接口0122相连,用来实现程序的在线调试;以太网接口0213与上位机01的以太网接口0123进行连接,从而实现arm9微处理器023与上位机01的互通信;rs
‑
232串口0214与上位机01的rs
‑
232串口0124进行连接,从而实现arm9微处理器023与上位机01的互通信。
55.存储单元022包括内存储单元0221、缓存单元0222及外存储单元0223;其中,内存储单元0221与缓存单元0222进行连接,负责完成稻瘟菌、水稻多组学数据的存储;缓存单元0222与内存储单元0221、预处理单元0241、网络构建单元0242同时进行连接,负责完成稻瘟菌关键srna识别的中间数据的存储;外存储单元0223与关键srna识别单元0244及rs
‑
232串口0214同时进行连接,负责完成稻瘟菌关键srna识别的结果数据的存储及通过rs
‑
232串口传回到上位机的显示单元上进行输出显示。
56.处理单元024包括预处理单元0241、网络构建单元0242、致病因子挖掘单元0243及关键srna识别单元0244;其中,网络构建单元封装了聚类降维算法(multi
‑
omics cluster
‑
dr),致病因子挖掘单元封装了多组学数据综合聚类(modsc)算法,关键srna识别单元,能够再致病因子网络中识别出稻瘟菌关键致病srna,并利用两物种样本的多元秩和检验方法(mrst)对识别出的关键致病srna进行聚类,为后续稻瘟菌
‑
水稻互作的研究提供支持。
57.在本实施例中,通过一台通用的pc计算机作为上位机01,该上位机可通过rs
‑
232串口和基于三星公司生产的32位的arm920t核的微处理器的基于多组学数据整合的稻瘟菌侵染水稻关键srna识别器的生成装置进行连接,共同作用以完成稻瘟菌关键srna识别的任务。
58.上位机01的输入单元0111及显示单元0131均采用pc计算机的输入及输出设备来实现其功能。
59.通过上位机01的以太网接口0123及arm9微处理器023的以太网接口0213实现上位机01与arm9微处理器023的互通信,以太网接口采用dm9000完全综合的、成本较低的单一快速以太网控制器芯片。
60.同时,增加了上位机01的jtag调试接口0122及arm9微处理器023的jtag调试接口0212,将此类接口通过jtag仿真(编程器)转换设备进行连接,可以实现上位机01实时地对arm9微处理器023上程序的分析和执行的监控。
61.usb接口采用的是usb3.0接口,为了实现存储单元的扩增,可以将稻瘟菌关键srna的识别结果数据通过上位机01的usb接口0121或arm9微处理器023的usb接口0211转存到u盘。
62.arm9微处理器023系统程序存储单元022选用的是32m hynix公司的hy57v561620ct sdram作为内存储单元0221,64m samsung公司的k9f1208uom nand flash作为缓存单元0222,及100g的硬盘作为扩展外存储单元0223。
63.arm9微处理器023的处理单元024中所包含的各单元均是封装在arm9微处理器上的稻瘟菌关键srna识别的深度学习算法,并且在运算时使用32位运算部件。
64.如图2所示,为稻瘟菌侵染水稻关键srna识别器的模型原理图,其连接关系如下:usb接口0211、jtag调试接口0212、以太网接口0213及rs
‑
232串口0214的数据输入口vin分别与arm9微处理器023的数据输出引脚vout1[0..7]相连,其gnd分别与arm9微处理器023的
gnd相连。
[0065]
内存储单元0221的数据输入口vin与arm9微处理器023的数据输出引脚vout1[0..7]相连,其数据输出口vout与缓存单元0222的数据输入口vin相连,其gnd与arm9微处理器023的gnd相连。缓存单元0222的数据输入口vin与内存储单元0221的数据输出口vout相连,其数据输出口vout与预处理单元的数据输入口、网络构建单元的数据输入口vin以及arm9微处理器023的数据输入引脚vin1[0..7]相连,其gnd与arm9微处理器023的gnd相连。外存储单元0223的数据输入口vin与关键srna识别单元0244的数据输出口vout相连,数据输出口vout与arm9微处理器023的数据输入引脚vin1[0..7]相连,gnd与arm9微处理器023的gnd相连。
[0066]
预处理单元0241的数据输入口vin与缓存单元0223的数据输出口vout相连,数据输出口vout与分别与arm9微处理器023vin1[0..7]及网络构建单元0242的数据输入口vin相连,gnd与arm9微处理器023的gnd相连。网络构建单元0242的数据输入口vin与预处理单元0241的数据输出口vout和缓存单元的数据输出口vout相连,数据输出口vout与致病因子挖掘单元0243的数据输入口vin相连,gnd与arm9微处理器023的gnd相连。致病因子挖掘单元0243的数据输入口vin与网络构建单元0242的数据输出口vout相连,数据输出口vout与关键srna识别单元数据的输入口vin相连,gnd与arm9微处理器023的gnd相连。关键srna识别单元0244的数据输入口vin致病因子挖掘单元0243的输出口vout相连,数据输出口vout与外存储单元0223的数据输入口vin相连,gnd与arm9微处理器023的gnd相连。
[0067]
如图3所示,稻瘟菌侵染水稻关键srna识别器电路图连接关系如下:
[0068]
1)f线为地址总线,分别连接arm9微处理器、外存储单元、缓存单元和内存单元的a0~a17引脚,负责传递地址信息;
[0069]
2)a线为数据总线,分别连接arm9微处理器、预处理单元、网络构建单元、致病因子挖掘单元、关键srna识别单元、内存储单元、缓存单元和外存储单元的d0~d15引脚,负责传输各种数据信息;
[0070]
3)b线为控制总线,分别连接arm9微处理器、外存储单元、缓存单元、内存储单元、预处理单元、网络构建单元、致病因子挖掘单元和关键srna识别单元的oe、wr、cs3、cs1、hbe和lbe引脚,负责传输控制信号和时序信号;
[0071]
4)c线为电源正极线,分别连接arm9微处理器、外存储单元、缓存单元、内存储单元、预处理单元、网络构建单元、致病因子挖掘单元、关键srna识别单元、usb接口、jtag接口、trs232接口和ds以太网接口的电源正极;
[0072]
5)d线为电源负极线,分别连接arm9微处理器、外存储单元、缓存单元、内存储单元、预处理单元、网络构建单元、致病因子挖掘单元、关键srna识别单元、usb接口、jtag接口、trs232接口和ds以太网接口的电源负极;
[0073]
6)e线为通用i/o线,分别连接arm9微处理器、usb接口、jtag接口和trs232接口,负责输入输出设备的数据传输;
[0074]
7)g线为网络接口线,分别连接arm9微处理器和ds以太网接口,负责网络传输。
[0075]
如图4所示,本发明还提供了一种基于多组学数据整合的稻瘟菌侵染水稻关键srna识别方法,包括如下步骤:
[0076]
(1)通过上位机的输入单元0111输入稻瘟菌、水稻多组学数据,并通过rs
‑
232串口
传至基于多组学数据整合的稻瘟菌侵染水稻关键srna识别器的内存储单元0221,并进一步将数据读到缓存单元0222中;
[0077]
(2)预处理单元0241从缓存单元0222中读取稻瘟菌、水稻多组学数据,进行预处理,并将结果输出到网络构建单元0242;
[0078]
(3)网络构建单元0242从预处理单元0241中读出预处理的多组学数据,处理后得到多组学分层异质互作网络;
[0079]
重复进行上述(2)~(3)操作直至所有的多组学数据均处理完成。
[0080]
建立多组学分层异质互作网络的过程包括:建立多组学分层异质横向网络构和纵向构建多组学分层异质网络。
[0081]
如图5所示,所述多组学分层异质横向网络构建过程如下:
[0082]
首先,在ncbi,mirbase,genebank,erice等多个数据库中收集稻瘟菌侵染水稻前后各组学数据并存储到本地数据库。
[0083]
然后分别在每个组学中建立横向关系网络:
[0084]
①
.基因组中,将由本地数据库中基因组学数据得到侵染前后的基因表达、拷贝数变异和dna甲基化数据分别用邻接矩阵x,d,c表示;使用基于关系数据的基因网络构建方法(gene
‑
relational data network)建立基因
‑
基因互作调控网络,完成基因组网络可视化。
[0085]
②
.转录组中,由本地数据库中的转录组学数据得到差异表达的srna;在starbase数据库中进行srna的靶基因预测并进行核心靶点选取,最终得到srna
‑
mrna互作网络。
[0086]
③
.蛋白组中,由本地数据库中的蛋白组学数据得到差异表达的蛋白质,将差异表达蛋白质输入到string数据库,利用其中的网络构建工具进行水稻ppi网络构建和稻瘟菌ppi网络构建;最后使用基于图的支持向量机方法(graph
‑
svm)完成节点选取和网络可视化。
[0087]
将网络中的正标签节点进行筛选并可视化蛋白质组学数据网络,建立稻瘟菌
‑
水稻蛋白组数据互作网络。
[0088]
④
.代谢组中,由本地数据库中的代谢组学数据得到差异表达的代谢数据,通过pca降维,再将水稻和稻瘟菌的代谢组数据分别进行kegg代谢通路分析,建立代谢组学关系网络。
[0089]
之后,纵向构建多组学分层异质网络,本发明采用基于多组学数据的聚类降维算法(multi
‑
omics cluster
‑
dr)对各个组学的分层网络纵向建立多组学稻瘟菌
‑
水稻分层异质互作网络。采用pca降维算法分别对基因
‑
基因互作调控网络、srna
‑
mrna互作网络、稻瘟菌
‑
水稻蛋白组数据互作网络和稻瘟菌
‑
水稻蛋白组数据互作网络降维,记为dr method集合;然后应用张量模型cp将数据矩阵进行分解并表示为因子矩阵,再应用判断离群值算法如绝对中位值偏差(median absolute deviation,mad)对样本矩阵中的离群值进行删除并选择前5%的dr模型,将聚类算法应用到dr算法中;接下来通过局部连续元准则lcmc进行质量评测,选择测评值最高的结果作为输出。最后对多个组学关系网络进行可视化,建立稻瘟菌
‑
水稻多组学分层异质网络。具体过程下:
[0090]
s1、在dr method中,应用cp模型将张量样本
×
样本
×
dr method分解成大小为n
×
p,n
×
p和m
×
p的三个因子矩阵,其中,所述前两个矩阵表示样本模式的分解,所述第三个矩阵表示dr方法模型;
[0091]
s2、通过绝对中位值偏差(mad)方法的剔除标准进行离群值判定及删除,所述剔除标准为:
[0092][0093]
s3、选取离群值前5%的dr方法模型,将k聚类方法应用到dr方法集合中以得到基于k聚类的dr方法组;
[0094]
s4、通过局部连续元准则lcmc(ql)进行质量评测,选择最高值作为输出。对多个组学关系结构进行可视化,得到稻瘟菌
‑
水稻多组学分层异质互作网络。
[0095]
(4)在致病因子挖掘单元中将多组学分层异质互作网络作为模型的输入,对网络中稻瘟菌srna节点做层次聚类,得到分层聚类树,如果某些srna在一个生理过程或不同组织中总是具有相类似的调控作用,那么我们有理由认为这些srna在功能上是相关的,可以把他们定义为一个模块。聚类树的不同分支代表不同的srna模块。将srna按照调控模式进行分类,将模式相似的srna归为一个模块。输出为致病srna调控模块,然后把致病srna调控模块传输给关键srna识别单元;
[0096]
(5)关键srna识别单元接收到致病srna调控模块,进行稻瘟菌关键srna识别,最终得出准确的识别结果;
[0097]
其中,在致病因子挖掘单元,将依次序读取多组学分层异质互作网络数据,致病因子挖掘单元将其作为模型的输入,将所有输出的致病srna调控模块传输到关键srna识别单元中;所有的数据处理完会得到识别结果,并将识别结果存储到外存储单元,通过rs
‑
232串口到达显示单元进行输出显示。
[0098]
在本实施例中,采用多组学数据综合聚类(modsc)算法建立稻瘟菌侵染水稻过程中的跨物种致病srna调控网络,揭示致病相关的各层组学因子之间的相互联系,挖掘稻瘟菌侵染水稻过程中的跨物种致病srna调控模块;并通过所述致病srna调控模块识别稻瘟菌致病相关的关键srna。如图6所示,modsc算法是局部双向聚类算法op
‑
cluster与全局多组学联合分析算法inmf结合的算法。
[0099]
首先构建初级致病调控网络,从构建完成的多组学分层异质网络中获取稻瘟菌srna
‑
水稻mrna数据矩阵,反复进行op
‑
cluster双聚类分析并挖掘致病因子,完成初级致病调控网络的构建。其中,op
‑
cluster双聚类分析算法是一种基于序列比对的双聚类算法,目的是发现具有一致的相对变化趋势的双聚类模块。为了对数据进行预处理,首先对每行的所有条目值按非递减顺序排序。其次,根据相似度将每个排序后的行组织成一个组序列,形如ba(cd),包括b、a、(cd)三个组,同组特征元素不分先后,其中(cd)内的列特征满足:|c
‑
d|≤0.1
×
min(c,d)。
[0100]
op
‑
cluster算法使用一个紧凑的树结构opc
‑
tree来存储用于挖掘op
‑
cluster的关键信息,同时发现频繁子序列和绑定与频繁子序列关联的行,其中共享相同前缀的序列将被收集并记录再同一位置。因此,对于共享前缀的所有行,沿着共享前缀的进一步操作将只执行一次,修剪技术也可以很容易地应用于opc树结构中,最终只需对opc
‑
tree进行一次dfs遍历即可得到全部的双聚类结果。
[0101]
然后构建完善致病调控网络,在多组学分层异质网络中获得多个组学之间生物标志物的内在联系,完整地保留了节点之间的结构关系,通过水稻mrna差异表达和稻瘟菌
srna变化选取出差异显著性(p
‑
value)小于0.05的致病相关的多组学数据输入到inmf多组学联合分析中,通过对op
‑
cluster构建的初级致病调控网络进行修剪从而完成完善致病调控网络的构建。其中inmf多组学联合分析方法如下,inmf联合拟合k个高斯联合潜在变量模型,在k个维度为p
k
*n的数据集x
k
(n为样本数)捕获共享和数据特定的结构。首先,用非负约束代替正交约束估计潜在变量,其次,稀疏矩阵h
k
在特定于数据的v
k
基矩阵和公共共享的w基矩阵之间共享,最后根据潜在因子e(w|x)的后验期望值进行k
‑
均值聚类确定聚类分配。其中系数矩阵和基矩阵是加载和潜在变量矩阵的对应矩阵。通过inmf聚类分析,进一步完善致病因子调控网络的构建。其中,inmf使用欧式损失函数优化问题,其模型公式为:
[0102][0103]
其中,w≥0,h
k
≥0,k=1,
…
,k。
[0104]
基于致病因子网络及modsc方法识别关键srna,过程包括:
[0105]
在致病因子网络中选取与稻瘟病表达最相关的前5%的srna调控模块,并从选取的srna调控模块中选取差异表达p
‑
value小于0.05的srna节点作为稻瘟菌致病相关的关键srna。
[0106]
若后续有需要利用识别出的关键srna做进一步研究,可利用两物种样本的多元秩和检验方法(mrst)对选取的关键致病srna进行聚类。
[0107]
上述两物种样本的多元秩和检验方法用于分析稻瘟菌srna高通量数据差异。首先仅选取在两组表达数据共有的srna从而在维度上进行统一,然后在组内进行数据的标准化从而在量上进行统一,可以得到标准化后的x1,x2,
…
,x
n
,其中x
i
代表第i个样本的观测值。对于x
i
,计算空间秩公式为:
[0108]
r
i
=m
j
[s(a
x
(x
i
‑
x
j
))],i,j=1,2,...,n
[0109]
其中,m[z]表示所有样本z的平均数,m
j
[z]表示以上平均数的第j维,s(z)表示z的符号(+或者
‑
),使a
x
满足其中λ为调节系数。
[0110]
选取两样本空间秩统计量为:其中为空间中心秩的平均向量,s=1,2,n
s
为权值。
[0111]
计算致病因子网络中选取出的稻瘟菌关键srna,将功能相近的关键srna做聚类处理。
[0112]
尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1