一种新型蓝色有机室温磷光材料的发光性能计算方法
1.本发明涉及蓝色有机室温磷光材料技术领域,具体涉及一种新型蓝色有机室温磷光材料的发光性能计算方法。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.oled又称为有机发光二极管、有机电激光显示,oled属于一种电流型的有机发光器件,是通过载流子的注入和复合而致发光的现象,发光强度与注入的电流成正比。有机室温磷光材料是一种在室温下电磁辐射和离子射线激发时能产生磷光的新型有机发光材料。
4.随着oled产业的进一步拓展,有机发光材料成为制约oled产业链的重要一环。原有的荧光材料存在激子利用率低,发光效率低的问题;原有的磷光材料虽然可以解决发光效率的问题,但其制备过程中往往需要重金属,这就导致使用磷光材料成本价格高昂。发明人发现,现有的有机磷光材料中,红色和绿色有机磷光材料的研究已经较为成熟,但最受关注的蓝色有机磷光材料在纯度、发光效率以及使用寿命方面都有待提高,并且蓝色有机磷光材料使用中会产生“烧屏”“暗斑”等问题。因此,蓝色有机室温磷光材料的设计和发展非常有必要。
技术实现要素:
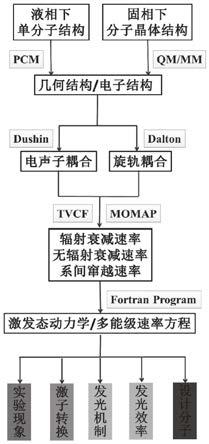
5.针对现有技术中的问题,本发明提供一种新型蓝色有机室温磷光材料的发光性能计算方法,该方法能够完善聚集态下聚集诱导室温磷光(aie
‑
rtp)材料的发光性能的计算方法,探究激发态动力学过程,揭示aie和rtp发光机制,明确分子聚集结构与发光性质之间的关系,平衡高效率与长寿命之间的关系,为高磷光效率和长磷光寿命的新型蓝色有机室温磷光材料的设计提供理论依据。
6.本发明的方法采用的计算机模拟软件为dushin程序包和momap程序包。
7.本发明的方法通过密度泛函理论(dft)和含时密度泛函理论(td
‑
dft)研究分子基态和激发态的几何结构和电子结构信息,计算得到分子的单重态和三重态能级结构,确认发生系间窜越过程的通道。进而基于二阶响应理论方法计算得到单重态和三重态之间的自旋轨道耦合系数。进一步,通过dushin程序包计算得到黄昆因子和重组能,并对其进行振动分析。基于上述计算的数据,通过爱因斯坦自发辐射方程和热振动关联函数方法计算激发态的辐射和无辐射衰减速率以及系间窜越速率,研究其激发态动力学过程并定量计算发光效率和发光时间,相应的计算都可以通过momap程序包实现。通过结合能级结构和激发态衰减速率,建立并求解多能级速率方程,阐述激子转换过程。此外,比较液相下和固相下t1与s0势能面的极小能量交叉点(mecp)信息,阐述激发态能量失活过程,并对比液相下和固相下磷光效率的变化,阐述聚集诱导有机室温磷光机制。
8.本发明提供一种新型蓝色有机室温磷光材料的发光性能计算方法,以多类同分异构的aie
‑
rtp分子为研究对象,利用密度泛函理论(dft)和含时密度泛函理论(td
‑
dft)研究其发光机制,进而从理论上设计筛选发光效率高的蓝色有机室温磷光材料。
9.具体的,本发明的一种新型蓝色有机室温磷光材料的发光性能计算方法具体包括以下步骤:
10.(1)搭建计算模型:以aie
‑
rtp分子为第一组分,分别选取中心分子周围半径为的球内分子为外层,当中心分子的能量随周围分子半径的增加而不再变化时,确定外层区域的范围,搭建收敛的pcm计算模型和qm/mm计算模型;
11.(2)在搭建的计算模型的基础上,计算得到aie
‑
rtp分子的单重态和三重态能级结构,确认发生系间窜越过程的通道;进而基于二阶响应理论方法计算得到单重态和三重态之间的自旋轨道耦合系数;进一步计算得到黄昆因子和重组能,并对其进行振动分析;
12.(3)基于步骤(2)计算的数据,通过爱因斯坦自发辐射方程和热振动关联函数方法计算激发态的辐射衰减速率和无辐射衰减速率以及系间窜越速率,得到其激发态动力学过程并定量计算发光效率和发光时间。
13.本发明的一种或多种实施例的有益效果是:
14.发展聚集态下分子发光性质的相关理论计算方法,并通过研究其激发态动力学过程,阐述聚集诱导发光和有机室温磷光机制,为优化和设计新型高效率且长寿命aie
‑
rtp分子提供理论依据;
15.完善了聚集态下aie
‑
rtp分子的发光性能的计算方法;
16.探究激发态动力学过程,揭示aie和rtp发光机制,明确分子聚集结构与发光性质之间的关系;
17.平衡高效率与长寿命之间的关系,为设计高性能aie
‑
rtp分子提供理论指导。
附图说明
18.构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
19.图1为具体研究的技术路线流程图;
20.图2是实施例1搭建的qm/mm计算模型示意图;
21.图3(a)是实施例1的t1态失活过程示意图;
22.图3(b)实施例1的分子间弱相互作用示意图;
23.图4是实施例1的分子间弱相互作用能量分解图;
24.图5是实施例1的3种aie
‑
rtp分子结构示意图。
具体实施方式
25.应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。
26.本发明的新型蓝色有机室温磷光材料的发光性能计算方法包括以下三个方面:
27.(1)阐述同分异构效应和堆积效应对分子能级以及跃迁属性的影响
28.①
选取多类同分异构的aie
‑
rtp分子为研究对象,采用pcm方法来获取其液相下的分子几何结构和电子结构特征,进而研究其临位、间位、对位取代对分子能级,特别是s1和t1之间能级差的影响,阐述同分异构效应对分子能级的影响。同时定量计算s1和t1的(π,π*)和(n,π*)轨道跃迁组成比例,阐述同分异构效应对跃迁属性的影响,进而揭示分子结构与性质之间的关系。
29.②
通过qm/mm方法来研究分子固相下的基本属性,分析几何构型变化对分子间堆积方式的影响。同时采用独立梯度模型(igm)方法形象化地展示分子间的相互作用,并通过能量分解方法定量地衡量分子间相互作用的强弱,阐述不同堆积方式下分子间相互作用的变化情况,分析这种变化对轨道跃迁属性和能级的影响,进而揭示堆积效应对分子激发态属性的影响。
30.(2)探究激发态动力学过程并揭示aie
‑
rtp发光机制
31.通过第(1)部分得到了分子液相下和固相下的基本属性以及单重态和三重态之间的能级差;进而通过dushin程序包计算得到黄昆因子和重组能,并对其进行振动分析,阐述分子从液相到固相下无辐射能量损失途径的变化情况。此外,通过dalton程序包计算得到单重态和三重态之间的自旋轨道耦合系数,通过tvcf方法计算得到激发态衰减速率常数和系间窜越速率,结合能级结构分析其激发态动力学过程。通过对比液相下和固相下辐射速率、无辐射速率以及系间窜越速率的变化情况,揭示aie和rtp发光机制。同时,根据具体的能级结构特点,建立并求解多能级速率方程,进而阐述其单重态激子和三重态激子的转换过程。
32.(3)理论设计新型高效率且长寿命aie
‑
rtp分子
33.目前报道的rtp分子,经常会采用含有孤对电子的o、n、s等元素来提供n轨道,以及结合二苯并噻吩等共轭单元来提供π轨道,从而实现对激发态(π,π*)和(n,π*)跃迁组成比例的调控,进而提高自旋轨道耦合效应,促进系间窜越过程。根据前文所述,不仅可以通过调控激发态属性,减小单重态和三重态(s
‑
t)的能差也能有效地促进系间窜越过程。所以,从分子结构设计的角度出发,可以采用供体
‑
受体(d
‑
a)型结构来有效地分离最高占据轨道(homo)和最低未占据轨道(lumo),从而实现减小能差的目的。同时还可以对供体和受体进行基团修饰来调控s
‑
t能差。但s
‑
t能差也不能太小,避免产生tadf现象(能差通常小于0.1ev),降低长余辉aie
‑
rtp分子的激子利用率。其次,从改变分子聚集方式来抑制无辐射能量损失的角度出发,可以采用多种分子间相互作用来构建类似于金属有机框架(mofs)的超分子骨架结构,从而抑制分子基团的转动和振动运动等效应导致的无辐射能量损失,提高分子的发光效率。除此之外,构建聚合物rtp发光分子也是一种非常有效的设计思路。通过上述方法,对实验合成报道的rtp分子进行供体和受体基团的取代,进一步通过分子设计来构建超分子骨架或有机聚合物,进而优化设计新型高性能aie
‑
rtp分子。理论计算其磷光效率和磷光寿命,为合成制备高效率且长寿命aie
‑
rtp分子提供参考依据。
34.本发明的一种实施方式中,提供一种新型蓝色有机室温磷光材料的发光性能计算方法,包括以下步骤:
35.(1)搭建计算模型:以aie
‑
rtp分子为第一组分,分别选取中心分子周围半径为的球内分子为外层,当中心分子的能量随周围分子半径的增加而不再变化时,确
定外层区域的范围,搭建收敛的pcm计算模型和qm/mm计算模型;
36.(2)在搭建的计算模型的基础上,计算得到aie
‑
rtp分子的单重态和三重态能级结构,确认发生系间窜越过程的通道;进而基于二阶响应理论方法计算得到单重态和三重态之间的自旋轨道耦合系数;进一步计算得到黄昆因子和重组能,并对其进行振动分析;
37.(3)基于步骤(2)计算的数据,通过爱因斯坦自发辐射方程和热振动关联函数方法计算激发态的辐射衰减速率和无辐射衰减速率以及系间窜越速率,得到其激发态动力学过程并定量计算发光效率和发光时间。
38.优选的,中心分子周围半径选取或
39.优选的,所述pcm计算模型用来获取液相下的分子几何结构和电子结构特征;
40.优选的,所述qm/mm计算模型用来获取固相下的分子几何结构和电子结构特征;
41.优选的,步骤(2)中是通过密度泛函理论(dft)和含时密度泛函理论(td
‑
dft)来研究aie
‑
rtp分子基态和激发态的几何结构和电子结构信息;
42.优选的,所述aie
‑
rtp分子的结构式为:
[0043][0044]
优选的,步骤(2)中,通过dushin程序包计算得到黄昆因子和重组能。
[0045]
优选的,步骤(2)中,通过dalton程序包计算得到单重态和三重态之间的自旋轨道耦合系数。
[0046]
优选的,步骤(3)中,还包括通过结合能级结构和激发态衰减速率,建立并求解多能级速率方程的步骤,得到单重态激子和三重态激子的转换过程。
[0047]
优选的,步骤(3)中,通过tvcf方法计算得到激发态的辐射衰减速率和无辐射衰减速率以及系间窜越速率。
[0048]
优选的,步骤(3)中,定量计算发光效率和发光时间,相应的计算都通过momap程序包实现。
[0049]
下面结合附图和实施例对本发明进一步说明。
[0050]
实施例1
[0051]
该实施例基于密度泛函理论(dft)和含时密度泛函理论(td
‑
dft)研究了aie
‑
rtp的分子激发态动力学研究过程:
[0052]
1、以附图5中的三种aie
‑
rtp分子为例,搭建如附图2所示的计算模型:分别选取中心分子周围半径为以及等的球内分子为外层,研究外层分子对中间分子的影响,当中心分子的能量随所选取研究的周围分子半径的增加而不再变化时,
确定外层区域的范围,最终搭建收敛的qm/mm计算模型(如图2所示);
[0053]
2、通过密度泛函理论(dft)和含时密度泛函理论(td
‑
dft)研究分子基态和激发态的几何结构和电子结构信息,计算得到分子的单重态和三重态能级结构,确认发生系间窜越过程的通道。进而基于二阶响应理论方法计算得到单重态和三重态之间的自旋轨道耦合系数。进一步,通过dushin程序包计算得到黄昆因子和重组能,并对其进行振动分析。基于上述计算的数据,通过爱因斯坦自发辐射方程和热振动关联函数方法计算激发态的辐射和无辐射衰减速率以及系间窜越速率,得到其激发态动力学过程并定量计算发光效率和发光时间,相应的计算都通过momap程序包实现。通过结合能级结构和激发态衰减速率,建立并求解多能级速率方程,得到激子转换过程。此外,比较液相下和固相下t1与s0势能面的极小能量交叉点(mecp)信息(如图3a所示),阐述激发态能量失活过程,并对比液相下和固相下磷光效率的变化,得到聚集诱导有机室温磷光机制。
[0054]
通过独立梯度模型方法形象化地考察不同堆积方式下的分子间相互作用(如图3b,图4所示),图3(b)显示了分子间弱相互作用,图4为分子间弱相互作用能量分解图;并基于分子力场的方式进行能量分解分析,定量地衡量分子间相互作用的强弱,得到不同堆积方式下分子间相互作用的变化情况,相应的计算通过开源的multiwfn程序完成。通过分别对比液相下分子发光性质的变化、固相下分子发光性质的变化以及对比液相和固相下分子发光性质的变化,得到分子几何结构的变化以及堆积方式的改变对分子发光性质的影响,进而根据分子几何结构的变化以及堆积方式的改变对分子发光性质的影响设计得到高效率且长寿命aie
‑
rtp分子。
[0055]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1