一种低深度WGS下机数据的处理方法与流程
一种低深度wgs下机数据的处理方法
技术领域
1.本技术属于基因检测技术领域,更具体地说,它涉及一种低深度wgs下机数据的处理方法、建立基因组杂合性缺失loh的计算方法、建立大片段迁移lst的计算方法和端粒等位基因不平衡tai的计算方法。
背景技术:
2.dna双链断裂(double strand breaks)是一种dna损伤类型,严重时会导致染色体的断裂和重排等,由于没有互补链进行修复,所以dna序列难以恢复,造成遗传信息丢失,这种dna双链断裂需要同源重组修复。若同源重组修复能力缺失即发生hrd,则会导致基因组丧失稳定性,在基因组不稳定的情况下容易积累dna损伤,以此恶性循环,导致癌症发生。hrd对铂类或者parp抑制剂的使用具有很重要的指导意义。
3.hrd一般由同源重组修复通路中基因变异或者表观变异引起,同源重组修复通路中包含brca1/2、rad52/rad22、palb2、rad51家族、brip1/bach1、atm和chek2等基因。研究表明存在brca1突变的女性,会存在50-85%和15-45%的概率罹患乳腺癌和卵巢癌。在乳腺癌中,遗传性brca1/2变异占大约7%,而在三阴性乳腺癌中能达到11%-15%。在家族性和偶发性乳腺癌患者中,估计有40%属于同源重组缺陷。尽管目前主要关注hrd在乳腺癌中的治疗,但是hrd在其他癌种中也是一个重要的指标。
4.目前,hrd的检测方法有以下两种:hr基因芯片,芯片设计时包含同源重组通路基因,利用靶向捕获技术和二代测序技术,获得同源重组通路基因的测序数据,检测所有基因的snv、indel和large arrangement,缺点是可能会高估hrd,而且基于芯片检测时,芯片上的snp位点是固定的,只能检测特定位点的变异,具有一定的局限性。
5.全基因组测序(wgs),对全基因组进行测序,检测染色体结构变异:包括杂合性缺失-loh,端粒位点不平衡-tai和大程度基因组不稳定性-lst来计算hrd score。优点是准确率高;缺点是成本相对较高。
技术实现要素:
6.为了在保证灵敏度及准确率的情况下降低成本,本技术提供一种基于低深度wgs评估hrd score的方法,该方法以低深度wgs测序所获得数据为基础进行评估hrd score,其降低了成本,更适合于大规模应用于临床。
7.本技术是通过以下方案实现的:本技术提供一种基于低深度wgs评估hrd score的方法,包括如下步骤:处理待测样本的低深度wgs下机数据;以及选自以下步骤中的任意一个或多个步骤:步骤一:建立基因组杂合性缺失loh的计算方法,获得hrd-loh score;步骤二:建立端粒等位基因不平衡tai的计算方法,获得hrd-tai score;和,步骤三:建立大片段迁移lst的计算方法,获得hrd-lst score。
8.本技术以低深度wgs下机数据为基础,建立评估hrd score的方法,大大降低了全基因组测序(wgs)的成本,相较于hr基因芯片检测,本技术检测的位点更加灵活,检测待测样本的结果更加准确,符合待测样本真实情况。
9.在本技术的一个具体实施方式中,所述处理待测样本的低深度wgs下机数据具体包括:s1-1:将所述下机数据与人类全基因组的参考基因组比对,得到第一比对文件;s1-2:去除所述第一比对文件中重复的reads,得到第二比对文件;s1-3:将人类全基因组划分成100kbp大小的windows。
10.本技术中,将全基因组按照顺序,以100kbp大小划分为不同的windows,便于后续数据的分析和处理。
11.在本技术的一个具体实施方式中,所述处理待测样本的低深度wgs下机数据还包括:s1-4:以所述第二比对文件中的reads为基本单元,统计落在每个window内的reads数,作为该window的reads count,记为rci,i为全基因组中按照排列顺序划分成的window的次序,i为1,2,3....;s1-5:统计每个window的gc碱基含量,将相邻的gc含量相同的windows合并为一组,第j组记为wj,第j组含有的window的个数记为mj,第j组含有的第k个window记为w
kj
,j、k分别为1,2,3...;s1-6:计算每个wj的中位值,记为rcj,与该样本整体的平均rc,记为rc
p
,通过以下公式对rci进行矫正:i=m1+m2+m3...+m
(j-1)
+k;s1-7:按照步骤s1-1、s1-2和s1-3处理n个健康样本的低深度wgs下机数据,计算每个window在n个健康样本中的中位值rc,记为rcy,作为该window的rc,构建baseline,n≥30,y为1,2,3...;s1-8:对待测样本的windows和健康样本的windows进行遍历,取每个window待测样本的nrci除以对应baseline中的rcy,得到dr;s1-9:基于循环二元分割算法(cbs)对dr进行分段,记为dr片段,同一个dr片段中的dr值比较接近,相邻两个dr片段的平均dr值相差显著,且每个dr片段中至少包含10个windows。
12.本技术中,设置每个dr片段中至少包含10个windows,其中10个windows可以保证在每个dr片段中保留长度在1m以上的片段,以最大可能的屏蔽掉干扰信号。
13.在本技术的一个具体实施方式中,所述处理待测样本的低深度wgs下机数据还包括:s1-10:统计每个dr片段中dr的中位值,作为该dr片段的dr值,记为drq,计算该dr片段的拷贝数,记为cq,计算公式为:。
14.本技术中,通过计算cq值,可以初步了解癌症发生的内在原因,可以在细胞学水平
上对症下药,使得癌症的缓解率大大提高。如果cq值不等于2,则说明发生了基因拷贝数的变异。cq值大于2意味着基因增加(gain),小于2则意味着基因缺失(loss)。如果某些负责细胞增殖的基因发生gain或者抑癌基因发生loss,则有可能引发细胞无限增殖,导致癌症的发生。因此,可以根据cq值初步判断癌症的发生。
15.在本技术的一个具体实施方式中,所述建立基因组杂合性缺失loh的计算方法具体包括:s2-1:使用千人基因组计划数据,选择杂合概率较高的snp位点;s2-2:统计所述snp位点上的每个位点等位碱基在待测样本上的频率,如果存在多个等位碱基,取频率最高的两个;如果仅有一个等位碱基,第二等位碱基给定默认频率为0;s2-3:统计每个window中所述snp位点第二等位碱基频率的平均数作为该window的af(allele frequency),生成新的af数列;如果af大于0,则将af调整成0.5;s2-4:将步骤s2-3中所述af相同的且相邻的window相连,得到较大的af片段;s2-5:选取cq大于等于1,且af等于0的af片段,如果该片段长度大于15mb,且小于其所在整个染色体的长度,则记为一个loh事件;s2-6:记录待测样本中的loh事件,记为hrd-loh score。
16.在本技术的一个具体实施方式中,杂合概率较高的snp位点,是指杂合概率大于0.2,这些位点在基因组上大致均匀分布,共约110000个snp位点。
17.在本技术的一个具体实施方式中,所述建立大片段迁移lst的计算方法具体包括:s4-1:步骤s1-9中获得的dr片段中,去除dr片段小于3mb的片段;s4-2:以单一的染色体为分析目标,依次将dr片段与染色体进行比对处理,将该染色体上相邻的cq相同的dr片段合并为一个大片段,记为drd,依次分析处理所有的染色体;s4-3:对drd进行统计,如果形成drd的两个相邻的dr片段的长度均大于10mb,且中间间隔小于3mb,则记为一个lst事件;s4-4:记录待测样本中的lst事件,记为hrd-lst score。
18.在本技术的一个具体实施方式中,所述建立端粒等位基因不平衡tai的计算方法具体包括:s3-1:使用千人基因组计划数据,选择杂合概率较高的snp位点;s3-2:统计所述snp位点上每个位点等位碱基的变异频率,得到变异频率最高的两个频率,第一等位碱基频率af1和第二等位碱基频率af2,根据以下公式计算每个位点的afr值(allele frequency ratio);如果某位点没有变异则afr值为0,去除该位点;s3-3:计算每个window的平均afr,作为该window的afr,记为afr
p
,如果某window的afr值均为0,则该window的afr
p
为0;s3-4:将afr
p
小于0.5的临近window进行合并,将afr
p
大于0.5的临近window进行合并,分别生成afr片段;s3-5:如果某个afr片段包含端粒,长度大于11mb,且afr
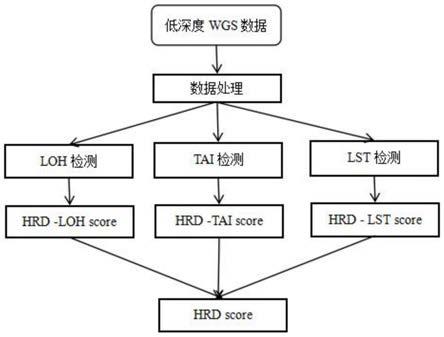
p
小于0.5,则记为一个tai事件;s3-6:记录待测样本中的tai事件,记为hrd-tai score。
19.在本技术的一个具体实施方式中,所述处理待测样本的低深度wgs下机数据还包括s1-0:下机数据的前处理:将下机数据去除reads上的接头。
20.在本技术的一个具体实施方式中,所述低深度wgs下机数据的文件格式为fastq格式。
21.在本技术的一个具体实施方式中,所述下机数据通过fastp软件去除reads上的接头。
22.在本技术的一个具体实施方式中,所述第一比对文件的格式为bam格式。
23.在本技术的一个具体实施方式中,所述下机数据与人类全基因组的参考基因组通过bwa软件进行比对。
24.在本技术的一个具体实施方式中,所述第一比对文件通过picard软件去除重复的reads,得到第二比对文件。
25.在本技术的一个具体实施方式中,所述第一比对文件在去除重复reads前,对第一比对文件进行碱基质量值矫正。
26.在本技术的一个具体实施方式中,所述第一比对文件通过gatk软件进行碱基质量矫正。
27.在本技术的一个具体实施方式中,所述hrd score = hrd-loh score + hrd-tai score + hrd-lst score。
28.在本技术的一个具体实施方式中,所述hrd阴性或阳性的cutoff值为hrd score=42。
29.在本技术的一个具体实施方式中,所述低深度wgs为10层以上的wgs测序结果。优选地,所述低深度wgs为10层的wgs测序结果。
30.本技术另一方面提供一种实现基于低深度wgs评估hrd score的装置,其包括:数据处理模块:用于处理低深度wgs下机数据;和选自以下统计模块中的一种或多种:hrd-loh score统计模块:用于判断并统计hrd-loh score;hrd-tai score统计模块:用于判断并统计hrd-tai score;和hrd-lst score统计模块:用于判断并统计hrd-lst score。
31.本技术提供的方法至少具有以下有益效果之一:本技术提供的基于低深度wgs评估hrd score的方法以低深度wgs测序形成的数据为基础进行分析,极大地降低了成本,有利于大规模的应用。
附图说明
32.图1为本技术实施例中提供的基于低深度wgs评估hrd score的方法流程示意图。
33.图2为本技术实施例中提供的不同hrd score患者的生存分析图。
34.图3为本技术实施例中提供的hrd score与brca1/2有害突变的关系图。
具体实施方式
35.除非另有定义,本技术中使用的所有技术和科学术语具有与本技术所述技术领域的普通技术人员通常理解的相同含义。
36.下面将结合本技术实施例,对本技术的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本技术一部分实施例,而不是全部的实施例。基于本技术中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本技术保护的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
37.本技术中缩略语和关键术语的定义如下:hr:homologous recombination,同源重组hrd:homologous recombination deficiency,同源重组修复缺陷loh:loss of hererozygosity,杂合性缺失tai:telomeric allelic imbalance,端粒位点不平衡lst:large-scale genomic instability,大程度基因组不稳定性hrd-loh score:长度大于15mb且小于染色体长度的loh事件个数hrd-tai score:延伸到染色体末端的位点不平衡,且区域长度大于11mb的事件个数hrd-lst score:染色体相邻的两个片段长度大于10mb,且两个片段之间距离小于3mb的事件个数本技术中,待测样本来源于公司接收的肿瘤患者样本,健康样本来源于公司收集的本公司员工贡献样本。全基因组来源于公开数据库ncbi,版本为hg19。千人基因组计划数据来源于公开数据库https://www.internationalgenome.org/data。
38.如图1所示,为本技术基于低深度wgs评估hrd score的过程。图1中是以hrd-loh score、hrd-tai score和hrd-lst score三者的总和评估hrd score。在实际应用过程中,可以根据hrd-loh score或者结合其他指标例如brca1/2突变等初步判断与杂合体缺失相关的疾病或指导用药等,也可以初步判断hrd score;可以根据hrd-tai score或者结合其他指标初步判断与端粒位点不平衡相关的疾病或指导用药等,也可以初步判断hrd score;以及根据hrd-lst score或者结合其他指标初步判断与大程度基因组不稳定性相关的疾病或指导用药等,也可以初步判断hrd score。
39.实施例1 低深度wgs数据处理的预处理1. 将低深度wgs下机数据通过fastp软件去除reads上的接头;2. 将处理后的下机数据与人类全基因组的参考基因组通过bwa软件进行比对,得到bam格式的第一比对文件;3. 将第一比对文件通过gatk软件进行碱基质量值矫正;4. 将矫正后的第一比对文件通过picard软件去除重复的reads,得到不包含重复reads的第二比对文件,文件的格式为bam格式。
40.5. 将人类全基因组按照排列顺序划分成100kbp大小的windows。
41.实施例2 构建dr片段1)以实施例1中的第二比对文件中的reads为基本单元,统计落在实施例1中每个window内的reads数,作为该window的reads count,记为rci,i为全基因组中按照排列顺序划分成的window的次序,i为1,2,3....。
42.2)统计每个window的gc碱基含量,将gc含量相同的相邻的windows合并为一组,第
j组记为wj,第j组含有的window的个数记为mj,第j组含有的第k个window记为w
kj
,j、k分别为1,2,3....;3)计算wj的中位值rc,记为rcj,与该待测样本整体的平均rc,记为rc
p
,通过以下公式对rci进行矫正:i=m1+m2+m3...+m
(j-1)
+k;4)按照步骤1)、2)和3)中的方法处理30个健康人低深度wgs数据,计算每个window在多个样本中的中位rc,记为rcy,作为该window的rc,构建baseline;n≥30,y为1,2,3...。
43.5)取每个window的待测样本的nrci除以对应baseline中的rcy,得到dr(depthratio);6)基于循环二元分割算法(cbs:circular binary segmentation)对dr进行分段(segment),记为dr片段,同一个dr片段中的dr值比较接近,相邻两个dr片段的平均dr值相差显著,且每个dr片段中至少包含10个windows。
44.实施例3 计算拷贝数统计每个dr片段中dr的中位值,作为该dr片段的dr值,记为drq,计算该dr片段的拷贝数,记为cq,计算公式为:。
45.本实施例中,可以通过cq(拷贝数)的计算,初步了解患者发生癌症的内在原因。
46.实施例4 hrd-loh score统计1. 使用千人基因组计划数据,选择杂合概率较高的snp位点,这些位点在基因组中大致均匀分布,约为110000个;2. 统计snp位点上的每个位点等位碱基在待测样本上的频率,如果存在多个等位碱基,取频率最高的两个;如果仅有一个等位碱基,第二等位碱基给定默认频率为0;3. 统计实施例1中每个window中snp位点第二等位碱基频率的平均数作为该window的af(allele frequency),生成新的af数列;如果af大于0,则将af调整成0.5;4. 将步骤3中,af相同且相邻的window相连,得到较大的af片段;5. 选取实施例3中的cq大于等于1,且af等于0的af片段,如果该片段长度大于15mb,且小于其所在整个染色体的长度,则记为一个loh事件;6. 记录待测样本中的loh事件,记为hrd-loh score。
47.实施例5 hrd
ꢀ‑ꢀ
tai score的统计1. 使用千人基因组计划数据,选择杂合概率较高的snp位点;2. 统计snp位点上每个位点等位碱基的变异频率,得到变异频率最高的两个频率,第一等位碱基频率af1和第二等位碱基频率af2,根据以下公式计算每个位点的afr值;如果某位点没有变异则afr值为0,去除该位点;3. 计算实施例1中每个window的平均afr,作为该window的afr,记为afr
p
,如果某window的afr值均为0,则该window的afr
p
为0;
4. 将afr
p
小于0.5的临近window进行合并,将afr
p
大于0.5的临近window进行合并,分别生成afr片段;5. 如果某个afr片段包含端粒,长度大于11mb,且afr
p
小于0.5,则记为一个tai事件;6. 记录待测样本中的tai事件,记为hrd-tai score。
48.实施例6 hrd
ꢀ‑ꢀ
lst score的统计1. 将实施例2中构建的dr片段中,去除dr片段小于3mb的片段;2. 以单一的染色体为分析目标,依次将dr片段与染色体进行比对处理,将该染色体上相邻的cq相同的dr片段合并为一个大片段,记为drd,依次分析处理所有的染色体;3. 对drd进行统计,如果形成drd的两个相邻的dr片段的长度都大于10mb,且中间间隔小于3mb,则记为一个lst事件;4. 记录待测样本中的lst事件,记为hrd-lst score。
49.实施例7 hrd score的统计hrd score = hrd-loh score + hrd-tai score + hrd-lst score。
50.本实施例中,hrd的cutoff值为hrd score=42,即hrd score大于42时,该患者对铂类药物和parp抑制剂敏感。
51.实施例8 低深度wgs测序层数的确定使用10个样本进行wgs测序,得到50多层的数据,然后分别随机截取50x、30x、20x、10x、5x,使用实施例1-7中的方法检测hrd-loh score、hrd-tai score和hrd-lst score,并计算最终的hrd score,进而进行最低测序量评估。其实验结果见表1所示。
52.表1 不同深度测序的相关性从表1可知,以50x的数据量的检测结果作为参考,当使用30x、20x、10x数据量的时候,得到的hrd-score与50x数据量时的相关系数超过95%,层数越低,相关系数越差,因此本技术实施例中以超过相关性大于95%的10x作为低深度wgs测序的最低层数。
53.应用例1利用本技术实施例1-8中确定方法分析某乳腺癌患者样本。
54.乳腺癌患者:姓名吴某,性别女,年龄46,临床症状左侧乳腺浸润性癌,无其他病史。
55.利用本技术实施例1-8中确定方法计算该患者的hrd-loh score为1,hrd
ꢀ‑ꢀ
tai score为4,hrd
ꢀ‑ꢀ
lst score为44,总的hrd score为49,高于cutoff值,则判断为hrd阳性。判断该患者对铂类化疗药物或者parp抑制剂等有很好的响应,此结果与已有文献报道hrd-high和铂类药物治疗敏感的相关性相符。在临床上,给患者使用了铂类化疗药物,其无进展生存期(pfs)达到了13个月,说明利用本技术实施例1-6中确定的方法计算hrd score,并通过hrd score可以指导临床用药。
56.应用例2选取43例乳腺癌样本和72例卵巢癌样本(共115例样本)进行低深度全基因组测序,115例样本具有铂类药物化疗的预后信息(即对115例样本均采用铂类药物化疗)。利用本技术实施例1-8中的方法计算loh、tai、lst,获取hrd score。
57.根据hrd score数值,以cutoff值=42为临界点,将115例样本分为两组,hrd-high(阳性)组和hrd-low(阴性)组。其中,hrd-high组为40例,hrd-low组为75例。结合临床pfs(progress free survival),对hrd-high组和hrd-low组进行生存分析,实验结果如图2所示。
58.从图2中可以看出,hrd-high组(40例)的整体生存时间要显著长于hrd-low(75例)组,说明hrd-high组对铂类药物治疗敏感,与实际相符。
59.分别检测115例样本brca1/2基因上的snv、indel有害变异。其结果为:43例乳腺癌样本中,brca1/2突变样本13例,brca1/2野生型30例;72例卵巢癌样本中,brca1/2突变样本27例,brca1/2野生型样本45例,制作brca1/2-hrd分布图,其结果如图3所示。
60.由图3可以看出,发生brca1/2突变的样本中,大部分hrd值较高,属于hrd阳性,该类患者对铂类药物敏感;而brca1/2野生型中也有少比例的样本属于hrd-high,结合图2可知,即使是brca1/2野生型,但如果其hrd score较高,属于hrd阳性,则该患者对铂类药物也敏感,与实际相符。因此,利用本技术实施例中提供的方法计算hrd score,通过hrd score的数值,在一定程度上能够指导临床用药。
61.本具体实施例仅仅是对本技术的解释,其并不是对本技术的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本技术的权利要求范围内都受到专利法的保护。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1