尼群地平-吲哚美辛无定型耦合体系的制备及效果分析
1.本发明涉及一种采用无定型技术进行难溶性药物联合用药的制剂领域,具体是尼群地平-吲哚美辛无定型耦合体系的制备及效果分析。
背景技术:
2.在药物的不同发展阶段和领域,已经上市的40%药物和75%的候选药物的低溶解度一直是一个很大的挑战,药物低溶解度和低溶出速率可能会在口服给药后对其生物利用度有限制。改良bcsⅱ类药的溶出的技术常见的有水溶性盐的制备、表面活性剂的添加,包合物的制备、固体分散技术、纳米技术等。bcs ii类药物的生物利用度很低,他们中的大多数是晶体药物。晶体药物的分子特征表明,在排列和结构上有一定的顺序,具有较高的晶格能。在溶出和吸收过程中,必须克服晶体的晶格能,这严重限制了药物的生物利用度。
3.近年,新型无定型耦合体系的研究开始受到国内外药剂学研究人员的关注,无定型耦合体系又称共无定型,无定型耦合体系是将两种或多种药物通过特殊的制备工艺得到的具有单一玻璃化转变温度的单相无定型耦合体系。该体系不仅具有单无定型药物的各种优点,还可改善药物的溶解度,溶出速率,提高药物的生物利用度等。某些药物的共无定型较单个无定型药物具有更高的溶解度和溶出速率,并且还可克服单个无定型药物通常不稳定的特点。
4.尼群地平分子量为360.36,熔点为156-167℃,为黄色结晶或结晶性粉末,无臭无味,遇光易变质。在丙酮、氯仿中易容,在甲醇、乙醇中溶解,在水中几乎不溶。尼群地平为第二代二氢吡啶类钙离子通道拮抗剂,主要作用于外周血管,通过抑制血管平滑肌和心肌的跨膜钙离子内流,引起冠状动脉、肾小动脉等全身血管的扩张,外周阻力下降产生降压作用。尼群地平适用于各种类型的高血压治疗,尤其伴发高血脂症的高血压患者、伴有冠心病的高血压患者和充血性心力衰竭患者。由于其溶解度极低,生物利用度低,制备制剂存在一定的困难。
5.吲哚美辛(indomethacin)分子量为357.79℃,熔点为158~162℃,为白色或微黄色结晶型粉末,在丙酮中溶解,在乙醇、乙醚,氯仿略溶,在水中几乎不溶,无味,几乎无臭。吲哚美辛是最强的pg合成酶抑制药之一,对cox-1和cox-2均有强大的抑制作用,也能抑制磷脂酶a2和磷脂酶c,减少粒细胞游走和淋巴细胞的增值,其抗炎作用比阿司匹林强10-40倍。故有显著抗炎及解热作用,对炎性疼痛有明显镇痛效果。但不良反应多,故仅用于其他药物不能耐受或疗效不显著的病例。对急性风湿性及类风湿性关节炎,约三分之二患者可得到明显改善。这类药物具有高渗透和低溶解的特性,是目前制剂研发中所占制剂候选药物比例中最大的原料药物群体。
技术实现要素:
6.本发明的目的在于提供尼群地平-吲哚美辛无定型耦合体系,为实现上述目的,本发明提供如下技术方案,以解决上述背景技术中提出的问题。
7.尼群地平-吲哚美辛无定型耦合体系的制备及效果分析,方法为:
8.(1)单无定型体系的制备:a.分别称取尼群地平和吲哚美辛原料药,分别置于预热的坩埚中,密封油浴热熔5min;b.将密封坩埚用液氮淬冷;c.避光烘箱中,在35℃下烘干; d.将烘干后的药品研磨成粉末,分别过120目筛后密封避光保存。
9.(2尼群地平-吲哚美辛无定型耦合体系的制备:按照质量比1:1、1:2、2:1称取尼群地平和吲哚美辛原料药,置于预热的坩埚中,密封油浴热熔5min后,将密封坩埚用液氮淬冷;将坩埚置于避光烘箱中,在35℃下烘干;将烘干后的药品研磨成粉末,分别过120目筛后密封避光保存,得到三个不同配比尼群地平-吲哚美辛无定型耦合体系(1:1、2:1、1:2)。
10.(3)对尼群地平-吲哚美辛无定型耦合体系的效果进行分析:a.x-射线衍射实验确认尼群地平-吲哚美辛无定型耦合体系构建成功;b.取尼群地平-吲哚美辛无定型耦合体系样品一定条件下放置3个月后,测定其稳定性;c.扫描电镜试验分析尼群地平-吲哚美辛无定型耦合体系的药物形状及其表面性质;d.topem实验分析尼群地平-吲哚美辛无定型耦合体系的tg值变化;e.体外增溶试验测定尼群地平-吲哚美辛无定型耦合体系的药品含量、溶出速率、溶解度变化。
11.优选地,步骤(1)和步骤(2)中油浴加热熔融时,以恰好开始熔化的温度点做为加热温度,该处理方法下制备的样品无碳化现象出现,因此确定各组药品制备时的加热温度分别是:吲哚美辛原料药为180℃;尼群地平原料药为185℃;质量比为1:1的尼群地平
‑ꢀ
吲哚美辛共无定型耦合体系为185℃;质量比为2:1的尼群地平-吲哚美辛共无定型耦合体系为188℃;质量比为1:2尼群地平-吲哚美辛共无定型耦合体系为187℃。
12.优选地,对尼群地平-吲哚美辛无定型耦合体系的效果分析试验证明:尼群地平-吲哚美辛无定型耦合体系在室温、相对湿度为40%的环境中,较单无定型体系稳定;溶出率和溶解度均明显高于单无定型体系,且具有明显的结晶抑制效果。
13.优选地,通过对无定型耦合体系的效果分析,尼群地平-吲哚美辛无定型耦合体系的最优原料药配比质量比2:1。
14.与现有技术相比,本发明的有益效果是:
15.1.本发明采用尼群地平-吲哚美辛作为模型药物,通过采用熔融-淬冷的方法制备尼群地平-吲哚美辛无定型耦合体系,并采用一系列的实验分析手段对得到的无定型耦合体系的理化性质及效果进行分析对比,得到尼群地平-吲哚美辛无定型耦合体系的最优原料药配比为2:1,为尼群地平-吲哚美辛无定型耦合体系的联合用药提供了有力的数据基础。
16.2.本发明通过研究尼群地平-吲哚美辛无定型耦合体系的制备和和效果分析,为解决尼群地平和吲哚美辛单独用作临床治疗时,因其溶解度低而生物利用度低、制备药剂困难的技术难题提供了新的研发方向和有力的技术支撑,为该领域用药提供了新的方案。
附图说明
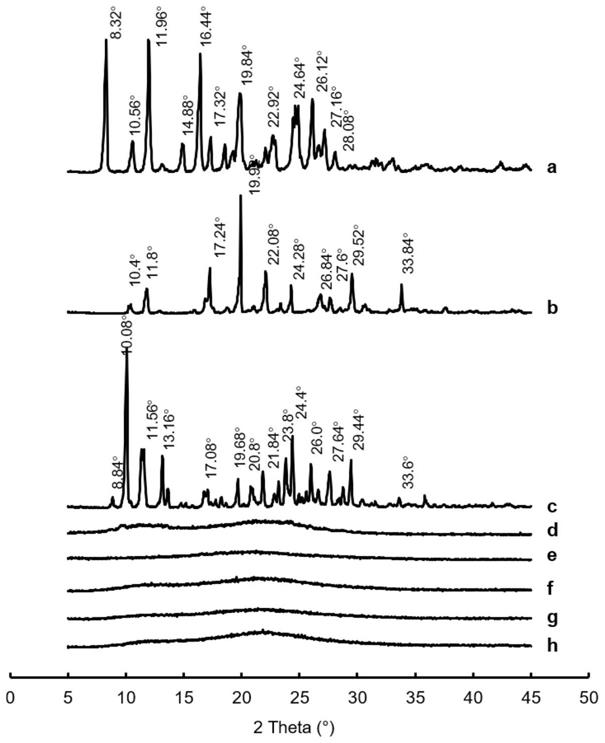
17.图1为测定各种样品x射线衍射图(图中a为尼群地平晶体,b为吲哚美辛晶体,c为尼群地平:吲哚美辛的物理混合物1:1,d为尼群地平单无定型e为吲哚美辛单无定型,f为尼群地平-吲哚美辛无定型耦合体系1:1,g为尼群地平-吲哚美辛无定型耦合体系1:2,h为尼群地平-吲哚美辛无定型耦合体系2:1);
18.图2为各种样品3个月稳定性x射线衍射图(a为尼群地平单无定型b为吲哚美辛单无定型,c为尼群地平-吲哚美辛无定型耦合体系1:1,d为尼群地平-吲哚美辛无定型耦合体系1:2, e为尼群地平-吲哚美辛无定型耦合体系2:1);
19.图3为电镜扫描各样品结构的结果对比图(图中a为尼群地平晶体,b为吲哚美辛晶体, c为尼群地平:吲哚美辛的物理混合物1:1,d为尼群地平单无定型,e为吲哚美辛单无定型, f为尼群地平-吲哚美辛无定型耦合体系1:1,g为尼群地平-吲哚美辛无定型耦合体系1:2,h 为尼群地平-吲哚美辛无定型耦合体系2:1);
20.图4为各无定型耦合体系样品及对照的tg值变化结果;
21.图5为检测各无定型耦合体系样品中尼群地平的含量变化结果;
22.图6为检测各无定型耦合体系样品中吲哚美辛的含量变化结果;
23.图7为各无定型耦合体系中及对照样品中尼群地平在0.25%sds水溶液中的溶出图;
24.图8为各无定型耦合体系及对照样品中吲哚美辛在0.1mol/l hcl中的溶出图;
25.图9为各无定型耦合体系中及对照样品中尼群地平在0.25%sds水溶液中24小时溶解度图;
26.图10为各无定型耦合体系及对照样品中吲哚美辛在0.1mol/l hcl中24小时溶解度图;
27.图11为不同浓度的吲哚美辛对尼群地平超饱和溶液的结晶抑制曲线图。
具体实施方式
28.以下的实施例便于更好的理解本发明,但并不限定本发明。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的试验材料,如无特殊说明,均为自常规生化试剂商店购买得到的。以下实施例中的定量试验,均设置三次重复试验,结果取平均值。
29.尼群地平-吲哚美辛无定型耦合体系的制备及效果分析,方法为:
30.(1)单无定型体系的制备:a.分别称取尼群地平和吲哚美辛原料药,分别置于预热的坩埚中,密封油浴热熔5min;b.将密封坩埚用液氮淬冷;c.避光烘箱中,在35℃下烘干; d.将烘干后的药品研磨成粉末,分别过120目筛后密封避光保存。
31.尼群地平单无定型体系的制备:称取尼群地平原料药500mg,置于已经预热的坩埚中,在油浴环境中加热至熔融状态,并在该状态下保持5min后,将小坩埚放在装满液氮的大坩埚中迅速骤冷(防止液氮中有杂质污染药品),该过程中要注意坩埚与锅盖的密封性,以防止药物与空气中骤冷的水蒸气接触,导致药物吸潮。另外,尼群地平遇光不稳定,制备过程中还应避光。熔融-淬冷后的尼群地平为黄色透明固体,固体状态在20到4℃下可较稳定的存在,将该样品储存于避光烘箱中,温度35℃,以烘干或减少样品中的水分,然后将样品置于研钵中研磨成粉末,过120目标准筛,取120目筛下细粉于自封袋中于避光干燥器中密封保存,备用待测。
32.吲哚美辛单无定型体系的制备过程同尼群地平单无定型体系的制备过程。
33.油浴熔融时,以恰好开始熔化的温度点做为加热温度,该处理方法下制备的样品无碳化现象出现,因此确定各组药品制备时的加热温度分别是:吲哚美辛原料药为180℃;
尼群地平原料药为185℃;质量比为1:1的尼群地平-吲哚美辛共无定型耦合体系(1:1)为 185℃;质量比为2:1的尼群地平-吲哚美辛共无定型耦合体系(2:1)为188℃;质量比为 1:2尼群地平-吲哚美辛共无定型耦合体系(1:2)为187℃。
34.(2尼群地平-吲哚美辛无定型耦合体系的制备:按照质量比1:1、1:2、2:1称取尼群地平和吲哚美辛原料药,置于预热的坩埚中,在油浴环境中加热至熔融状态,并在该状态下保持5min,后将小坩埚放在装满液氮的大坩埚中迅速骤冷(防止液氮中有杂质会污染药品),该过程中要注意坩埚与锅盖的密封性,以防止药物与空气中骤冷的水蒸气接触,导致药物吸潮;将坩埚置于避光烘箱中,在35℃下烘干;将烘干后的药品研磨成粉末,分别过120 目筛后密封避光保存,得到尼群地平-吲哚美辛无定型耦合体系(1:1、1:2、2:1)。
35.(2)对尼群地平-吲哚美辛无定型耦合体系(1:1、1:2、2:1)的效果进行分析:
36.a.x-射线衍射实验确认尼群地平-吲哚美辛无定型耦合体系构建成功:工作条件cuk α靶,电压40kv,电流40ma,扫描速度10
°
/min,扫描范围5-45
°
,将尼群地平晶体,吲哚美辛晶体,尼群地平与吲哚美辛1:1的物理混合物,尼群地平单无定型体系,吲哚美辛单无定型体系,尼群地平-吲哚美辛无定型耦合体系(1:1、1:2、2:1)分别进行x射线衍射。结果如图1所示。
37.b.取尼群地平-吲哚美辛无定型耦合体系(1:1、1:2、2:1)样品一定条件下放置3个月后,测定其稳定性:将尼群地平单无定型,吲哚美辛单无定型,尼群地平-吲哚美辛无定型耦合体系(1:1、1:2、2:1)放置在25℃,相对湿度为30%的环境中1个月、3个月、4 个月、5个月后测定其x射线衍射。结果如图2所示。
38.c.扫描电镜试验分析尼群地平-吲哚美辛无定型耦合体系(1:1、1:2、2:1)的药物形貌及其表面性质:利用sem电子扫描显微镜来观察药物的形状及其表面性质,分别对尼群地平晶体,吲哚美辛晶体,物理混合物1:1,尼群地平单无定型体系,吲哚美辛单无定型体系,尼群地平-吲哚美辛无定型耦合体系(1:1),尼群地平-吲哚美辛无定型耦合体系 (1:2),尼群地平-吲哚美辛无定型耦合体系(2:1)进行电子扫描,观察各组药物结构及变化。结果如图3所示。
39.d.topem实验分析尼群地平-吲哚美辛无定型耦合体系的玻璃化转变温度tg值变化:topem实验,氮气气流氛围,传感器左侧标有s,放置待测样品坩埚;右侧标有r,放置参比坩埚,扫描速度2℃/min,扫描范围20-150℃,进行尼群地平无定型,吲哚美辛无定型,尼群地平-吲哚美辛共无定型(1:1,1:2,2:1)dsc试验。通过gordon-taylor方程可以推算出无定型药物耦合体系的理论tg值,方程如下:
40.t
gab
=(wa·
t
ga
+k
·
wb·
t
gb
)/(wa+k
·
wb)
41.t
gab
代表无定型耦合体系的玻璃化转变温度,wa和wb分别代表药物组分a和b在无定型耦合体系中所占的重量百分数,t
ga
和t
gb
分别为单个无定型药物组分a和b的玻璃化转变温度。
42.其中k为常数,可由下列公式求得
43.k≈δc
pb
/δc
pa
44.δc
pa
和δc
pb
分别代表单个无定型药物组分在玻璃化转变温度变化前后的热容变化值。各种样品tg值变化见图4。
45.由图4可知tg实测值均与理论值呈正偏差,表明两种混合物在各比例的无定型耦
合体系中两组分之间氢键的数量和强度强于单一组分中存在的相互作用,有利于药物无定型耦合体系的形成。因此,通过比较实测tg值与理论tg值的大小可以推测无定型耦合体系中药物相互作用的强弱。综上所述,该实验结果可较深入的说明无定型耦合体系比单独无定型具有更强的稳定性,为揭示无定型耦合体系的稳定化特征及形成提供了新理论依据。
46.e.体外增溶试验测定尼群地平-吲哚美辛无定型耦合体系的药品含量、溶出速率、溶解度变化。
47.药品含量变化:准确称取各药品样品,分别在容量瓶中,用甲醇溶并稀释至刻度,定容,静置半小时,做n=3平行试验,用高效液相色谱仪测定其面积,计算含量。尼群地平、吲哚美辛在各无定型耦合体系中的含量结果分别如图5、6所示。经过淬冷过程,两种药物含量无明显下降,可以基本保持化学稳定。
48.溶出效率分析:称取各种样品于900ml介质(介质可为:0.25%sds水溶液、0.1mol/lhcl)中,采用桨法,在37℃,100r/min条件下,于5、10、15、30、45、60min时通过 0.45微米微孔滤膜取5ml,取20μl注入高效液相色谱仪,测定,相同条件重复3次,得其溶出曲线。结果如图7、图8所示。结果表明,尼群地平-吲哚美辛无定型耦合体系(2:1) 同时引起了两种药物的最大溶出。
49.溶解度变化分析:取各种样品加入到50ml比色管中,加入0.25%sds水溶液或 0.1mol/l hcl至50ml,在37℃恒温摇床中以100r/min的转速放置24小时取上清液过0.45 微米滤膜在尼群地平液相条件下进样测定,以上做n=3的平行试验,通过与对照组比较,分析尼群地平-吲哚美辛无定型耦合体系的溶解度变化。结果如图9、图10所示。结果表明,尼群地平-吲哚美辛无定型耦合体系(2:1)同时引起了两种药物的最大溶解度。
50.超饱和抑晶效果分析:称取尼群地平45mg于5ml ep管中,用最少量甲醇将其溶解。按1:0、1:1、1:2、1:3、1:4的比例分别称取吲哚美辛于5ml ep管中,用少量甲醇助溶后倒入于900ml ph8.0的磷酸盐缓冲介质中,采用桨法,在37℃,100r/min条件下,在30、60、90、120、150、180、210、240min时通过0.45微米微孔滤膜取5ml,取供试品20ul注入液相色谱仪,测定,相同条件重复3次,得其溶出曲线,结果作为对照组。结果如图11 所示。
51.以同样的方法测得尼群地平单无定型、吲哚美辛单无定型、尼群地平-吲哚美辛无定型耦合体系的溶出曲线,与对照组进行比较分析。
52.其中,对尼群地平-吲哚美辛无定型耦合体系的效果分析试验证明:尼群地平-吲哚美辛无定型耦合体系(1:1、1:2、2:1)在室温、相对湿度为40%的环境中,较单无定型体系较稳定;溶出率和溶解度均明显高于单无定型体系,且具有明显的结晶抑制效果。
53.其中,通过对无定型耦合体系的效果分析,尼群地平-吲哚美辛无定型耦合体系的最优原料药配比质量比2:1。
54.实施例一
55.x-射线衍射实验确认尼群地平-吲哚美辛无定型耦合体系构建成功:工作条件cukα靶,电压40kv,电流40ma,扫描速度10
°
/min,扫描范围5-45
°
,将尼群地平晶体,吲哚美辛晶体,尼群地平与吲哚美辛1:1的物理混合物,尼群地平单无定型体系,吲哚美辛单无定型体系,尼群地平-吲哚美辛无定型耦合体系(1:1、1:2、2:1)分别进行x射线衍射。结果如图1所示:尼群地平晶体,吲哚美辛晶体,尼群地平与吲哚美辛1:1的物理混合物均由尖锐而又明显的晶体特征峰,而尼群地平单无定型体系,吲哚美辛单无定型体系、尼群地平-吲哚美辛
无定型耦合体系(1:1、1:2、2:1)的晶体特征峰几乎全部消失,由此可推知经熔融-淬冷处理后的尼群地平-吲哚美辛各比例的无定型耦合体系有着更为彻底的非晶态化。综上可初步确定尼群地平无定型,吲哚美辛无定型,以及各比例的尼群地平-吲哚美辛无定型耦合体系成功构建。
56.实施例二
57.将尼群地平无定型,吲哚美辛无定型,尼群地平-吲哚美辛共无定型耦合体系(1:1, 1:2,2:1)放置在25℃,相对湿度为30%的环境中3个月后测定其x射线衍射结果。结果如图2所示:尼群地平-吲哚美辛无定型耦合体系(1:1,1:2,2:1)各比例较单无定型的晶体衍射峰均弱且小;由此可知,在固体状态下,尼群地平-吲哚美辛共无定型转晶的时间较单无定型转晶的时间要延后,因此,尼群地平-吲哚美辛共无定型较单无定型更加稳定,其中以尼群地平-吲哚美辛无定型耦合体系(1:2)出现的晶体峰最少,最为稳定。
58.实施例三
59.利用sem电子扫描显微镜来观察药物的形状及其表面性质,分别对尼群地平晶体,吲哚美辛晶体,物理混合物1:1,尼群地平单无定型体系,吲哚美辛单无定型体系,尼群地平-吲哚美辛无定型耦合体系(1:1),尼群地平-吲哚美辛无定型耦合体系(1:2),尼群地平-吲哚美辛无定型耦合体系(2:1)进行电子扫描,观察各组药物结构及变化。结果如图3所示:从图a与图b可以看出尼群地平与吲哚美辛晶体的微观形态分布均匀,具有明显的片层结构;从图c可以看出物理混合物的微观形态同时拥有两种药物的分布状况以及片层结构;从图d与图e中可以看出通过熔融-淬冷处理后的尼群地平和吲哚美辛晶体,在形态上发生了较大的变化,呈无规则状态,片层结构减少甚至消失,初步推测其为无定型状态。从图f,图g与图h中可以看出通过熔融-淬冷处理后的无定型耦合体系各比例形态发生了更为明显的变化,片层结构几乎消失,无规则的排列顺序,从其微观形态上的变化可推知,两种药物之间发生了一定的相互作用,这一结果为新型无定型耦合体系的成功构建提供了又一可靠的依据。
60.实施例四
61.topem实验分析尼群地平-吲哚美辛无定型耦合体系的玻璃化转变温度tg值变化;利用温度调制型的dsc仪及topem实验进行测定分析。由图4可知,tg实测值均与理论值呈正偏差,表明两种混合物在各比例的无定型耦合体系中两组分之间氢键的数量和强度强于单一组分中存在的相互作用,有利于药物无定型耦合体系的形成。因此,通过比较实测tg值与理论tg值的大小可以推测无定型耦合体系中药物相互作用的强弱。综上所述,该实验结果可较深入的说明无定型耦合体系比单独无定型具有更强的稳定性,为揭示无定型耦合体系的稳定化特征及形成提供了新理论依据。
62.实施例五
63.药品含量变化:准确称取各药品样品,以尼群地平为参照,相当于尼群地平25mg,分别在50ml容量瓶中,用甲醇溶并稀释至刻度,定容,静置半小时,做n=3平行试验,用高效液相色谱仪测定其面积,计算含量。结果如图5、图6所示:经熔融-淬冷处理后的各样品中尼群地平的含量损失较小,所以,该系统的构建并未对其药物含量产生较大波动。
64.实施例六
65.称取尼群地平50mg,各种样品(相当于尼群地平50mg),于900ml 0.25%sds的蒸馏
水中,采用桨法,在37℃,100r/min条件下,于5、10、15、30、45、60min时通过0.45 微米微孔滤膜取5ml,取20μl注入高效液相色谱仪,测定,相同条件重复3次,得其溶出曲线。结果如图7所示:从图中可看出0~60min内,尼群地平-吲哚美辛共无定型(1:1, 1:2,2:1)组的溶出量均高于吲哚美辛晶体组以及尼群地平-吲哚美辛物理混合物(1:1)组;其中以尼群地平-吲哚美辛共无定型(2:1)组为最高,因此,在该条件下,尼群地平-吲哚美辛新型无定型耦合体系相对于晶体药物以及单组分药物无定型有明显的的增溶优势。
66.实施例七
67.称取吲哚美辛50mg,各种样品(相当于吲哚美辛50mg),于900ml0.1mol/l hcl中,采用桨法,在37℃,100r/min条件下,于5、10、15、30、45、60min时通过0.45微米微孔滤膜取5ml,取20μl注入高效液相色谱仪,测定,相同条件重复3次,得其溶出曲线。结果如图8所示:从图中可看出0~60min内,吲哚美辛-尼群地平共无定型组(1:1,1:2, 2:1)与吲哚美辛无定型组的溶出量均高于吲哚美辛晶体组以及吲哚美辛-尼群地平物理混合物1:1组;其中以尼群地平-吲哚美辛共无定型(2:1)组为最高,最大浓度可达到 19.29μg/ml;因此,在该条件下,尼群地平-吲哚美辛新型无定型耦合体系在增溶方面更具优势。
68.实施例八
69.取50mg的尼群地平,各种样品(相当于尼群地平50mg),加入到50ml比色管中,加入蒸馏水至50ml,在37℃恒温摇床中以100r/min的转速放置24小时取上清液过0.45微米滤膜在尼群地平液相条件下进样测定,以上做n=3的平行试验,通过与对照组比较,分析尼群地平-吲哚美辛无定型耦合体系的溶解度变化。结果如图9所示:尼群地平-吲哚美辛共无定型无定耦合体系部分比例增溶优势比较显著,尼群地平-吲哚美辛无定型耦合体系 (2:1)组浓度最高,据此可见共无定型耦合体系相对于单组分无定型,不但具备明显的增溶优势,而且具有更好的抑晶效果。
70.实施例九
71.取50mg的吲哚美辛,各种样品(相当于吲哚美辛50mg),加入到50ml比色管中,加入0.1mol/l hcl至50ml,在37℃恒温摇床中以100r/min的转速放置24小时取上清液过 0.45微米滤膜在尼群地平液相条件下进样测定,以上做n=3的平行试验,通过与对照组比较,分析尼群地平-吲哚美辛无定型耦合体系的溶解度变化。结果如图10所示:相对于24 小时各比例共无定型浓度均有提高,尼群地平-吲哚美辛共无定型各组浓度均高于单组分无定型,尼群地平-吲哚美辛共无定型(2:1)组浓度最高,由此可见共无定耦合体系相对于单晶体和物理混合物而言,具备明显的增溶优势。
72.实施例十
73.称取尼群地平45mg于5ml ep管中,用最少量甲醇将其溶解。按1:0、1:1、1:2、1:3、 1:4的比例分别称取吲哚美辛于5ml ep管中,用少量甲醇助溶后倒入于900ml ph8.0的磷酸盐缓冲介质中,采用桨法,在37℃,100r/min条件下,在30、60、90、120、150、180、 210、240min时通过0.45微米微孔滤膜取5ml,取供试品20ul注入液相色谱仪,测定,相同条件重复3次,得其溶出曲线。结果如图11所示:在液态状态下,随着吲哚美辛浓度的增加,尼群地平的浓度越高,没有加吲哚美辛的尼群地平溶液浓度最低。由此可知,在超饱和状态下,一种药物可以抑制另一种药物从超饱和状态下的结晶,可以推测在无定型耦合体系中,一种药物可以抑制另一种药物的结晶,增加相互无定型状态的稳定化。从而为两种药物的联合
应用提供了有力的数据。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1