检测纯合缺失的方法和装置与流程
1.本发明涉及基因测序领域,具体而言,涉及一种检测纯合缺失的方法和装置。
背景技术:
2.肿瘤组织细胞成分结构复杂,除了肿瘤细胞,还包括基质细胞、免疫细胞、 成纤维细胞、脉管系统和细胞外基质等,共同构成肿瘤微环境。肿瘤纯度是指肿瘤组织中肿瘤细胞所占的比例。因此实际的肿瘤组织测序数据中往往包含未知比例的正常细胞的数据。
3.基因的纯合缺失(homozygous deletion,hd)是一类在临床上非常重要的结构变异,与多种肿瘤的预后、靶向药物的敏感性相关。可靠的hd检测结果可以为临床用药以及病情评估等提供十分重要的依据。
4.目前临床上没有专门的hd检测方法或者技术,hd的检测一般归类于基因的拷贝数变异(copy number variation,cnv)检测。cnv目前的常用检测平台和金标准是基因芯片(microarray),其他技术包括基于pcr、免疫组化的实验手段(如fish,ihc等)、高通量测序(ngs)等。基于实验的方法通常单次检测仅可覆盖一个基因,且检测结果灵敏度较低。基于基因芯片的方法只能检测特定的几个区域,而且正常组织的含量的多少也会影响对肿瘤cnv的计算。基于ngs的检测方法通常基于测序深度侧重于杂合缺失(loh)和cnv扩增的计算,无法区分真实的缺失还是测序未覆盖。
5.分析hd难度源于以下三个方面: (1)肿瘤细胞几乎总是与未知比例的正常细胞混合; (2)肿瘤细胞的实际dna含量, 由于总数和结构的染色体异常而未知; (3)肿瘤细胞群由于持续的亚克隆进化而可能是异质性的。 原则上,可以根据每个肿瘤细胞的dna质量的细胞学测量或单细胞测序方法, 通过重新排列相对数据来推断绝对拷贝数。 然而, 这样的方法并不适合在解读肿瘤基因组中大规模使用。(4)对于测序深度为0的区域难以区分是未捕获到数据还是发生了纯合缺失。
6.因此,亟需研发一种更有效的准确识别肿瘤纯合缺失的方案,以满足科研和/或临床的使用需求。
技术实现要素:
7.本发明的主要目的在于提供一种检测纯合缺失的方法和装置,以解决现有难以区分某些缺失是否为纯合缺失的问题。
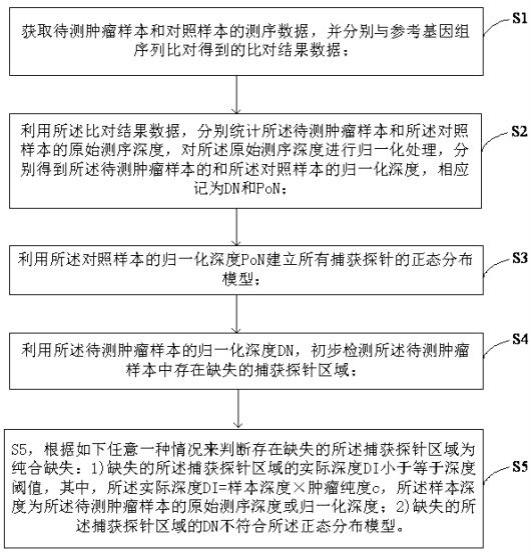
8.为了实现上述目的,根据本发明的一个方面,提供了一种检测纯合缺失的方法,该方法包括:s1,获取待测肿瘤样本和对照样本的测序数据,并分别与参考基因组序列比对得到的比对结果数据,配对样本包括待测肿瘤样本和对照样本;s2,利用比对结果数据,分别统计待测肿瘤样本和对照样本的原始测序深度,对原始测序深度进行归一化处理,分别得到待测肿瘤样本的和对照样本的归一化深度,相应记为dn和pon;s3,利用对照样本的归一化深度pon建立所有捕获探针的正态分布模型;s4,利用待测肿瘤样本的归一化深度dn,初步检测待测肿瘤样本中存在缺失的捕获探针区域;s5,根据如下任意一种情况来判断存在
缺失的捕获探针区域为纯合缺失:1)缺失的捕获探针区域的实际深度di小于等于深度阈值,其中,实际深度di=样本深度
×
肿瘤纯度c,样本深度为待测肿瘤样本的原始测序深度或归一化深度;2)缺失的捕获探针区域的dn不符合正态分布模型。
9.进一步地,s2包括:s21,按照如下原则统计捕获区域的原始测序深度:a) read1和read2的重叠区域只统计一次,b) 发生插入缺失的reads纳入统计;s22,在样本内,以捕获探针区域为单位对原始测序深度进行第一次归一化处理,得到样本内归一化深度;s23,在样本间,对同一捕获探针区域的样本内归一化深度进行第二次归一化处理,得到同一捕获探针区域的归一化深度;s24,将待测肿瘤样本的和对照样本的归一化深度分别记为dn和pon;优选地,第一次归一化处理通过每一捕获探针区域的原始测序深度/样本内所有捕获探针区域的原始测序深度的中位值得到;优选地,第二次归一化处理通过同一捕获探针区域的样本内归一化深度/所有样本在同一捕获探针区域的样本内归一化深度的中位值得到。
10.进一步地,在进行第一次归一化处理之前,方法还包括:去除原始测序深度为0的捕获探针区域;优选地,在第一次归一化处理和/或第二次归一化处理后,方法还包括将待测肿瘤样本中归一化后不满足正态分布模型的捕获探针区域作为备选分析区;优选地,若缺失的捕获探针区域位于备选分析区中,则推断存在缺失的捕获探针区域为纯合缺失。
11.进一步地,s4包括:以单个捕获探针区域为单位,检测待测肿瘤样本的归一化深度dn是否满足dn<待测肿瘤样本的归一化深度dn的均值
‑ꢀ
n*标准差,1.7≤n≤2.4,若是,判定捕获探针区域存在缺失。
12.进一步地,s5中,肿瘤纯度c按照如下方法计算:1)选取对照样本中0.15≤af≤0.85的杂合snp,从待测肿瘤样本中找出相应杂合snp的突变频率;2)以捕获探针区域为单位,从归一化处理后的测序数据中读取各杂合snp的支持read数,对对照样本中各杂合snp的af进行矫正,得到矫正后的af,其中,若双端测序的read 1和read 2都支持同一杂合snp,则只计算一次;3)以捕获探针区域为单位,分别统计对照样本和待测肿瘤样本的各个捕获探针区域的平均的归一化深度,并进一步分别计算对照样本和待测肿瘤样本的同一捕获探针区域内所有snp位点的归一化深度的方差,如果待测肿瘤样本的捕获探针区域内所有snp位点的归一化深度的方差超出对照样本在同一捕获探针区域的方差波动范围,则标记为异常区域;4)在异常区域中,将待测肿瘤样本的杂合snp中相对于对照样本的杂合snp位点中基因频率下降的等位基因记为caf,若矫正后的af≥0.5,则caf=1
‑
矫正后的af;若矫正后的af<0.5,则caf=矫正后的af;5)计算异常区域中,各捕获探针区域的log2(dn/pon)值,若捕获探针区域的log2(dn/pon)值<0,则表明捕获探针区域发生了杂合性缺失,此时肿瘤纯度c = (1
‑
2caf) / (1
‑
caf)。
13.进一步地,在计算各个捕获探针区域的平均的归一化深度时,如果相邻的捕获探针之间有重叠,则合并为一个捕获探针区域;优选地,在进行方差计算时,如果捕获探针区域所涵盖的物理距离大于等于1mb且待测肿瘤样本的方差超出对照样本在同一捕获探针区域的方差波动范围的,则将捕获探针区域进一步分割成多个,然后再进行方差的计算。
14.进一步地,获取待测肿瘤样本和对照样本的测序数据,并分别与参考基因组序列比对得到的比对结果数据包括:对待测肿瘤样本和对照样本的原始测序数据进行质控处理,得到有效数据;将有效数据与参考基因组序列进行比对,得到初步比对信息;对有效数
据中含有indel位点的区域进行重新比对,得到比对结果数据;优选地,重新比对包括:根据已知的indel位点信息,查找有效数据中所有的indel区域,形成indel区域序列文件;结合indel区域序列文件,对初步比对信息中的indel区域进行重新比对;优选地,质控处理包括: 根据碱基测序质量、reads长度、n碱基含量对原始测序数据进行筛选,同时去除原始测序数据中的测序引物序列和不能被测序引物序列识别的序列;优选地,待测肿瘤样本是前列腺癌石蜡包埋组织样本,原始测序数据是靶向捕获测序数据;更优选地,靶向捕获测序数据是靶向捕获如下14个同源重组修复基因的测序数据:brca1、brca2、atm、cdk12、palb2、 brip1、rad54l、bard1、rad51b、rad51d、chek1、chek2、fancl及rad51c。
15.为了实现上述目的,根据本发明的第二个方面,提供了一种检测纯合缺失的装置,该装置包括:获取模块,用于获取待测肿瘤样本和对照样本的测序数据,并分别与参考基因组序列比对得到的比对结果数据,配对样本包括待测肿瘤样本和对照样本;深度归一化模块,用于利用比对结果数据,分别统计待测肿瘤样本和对照样本的原始测序深度,对原始测序深度进行归一化处理,分别得到待测肿瘤样本的和对照样本的归一化深度,相应记为dn和pon;探针捕获特征模型构建模块:用于利用对照样本的归一化深度pon建立所有捕获探针的正态分布模型;缺失区域初检模块,用于利用待测肿瘤样本的归一化深度dn,初步检测待测肿瘤样本中存在缺失的捕获探针区域;纯合缺失推断模块,用于根据如下任意一种情况来推断存在缺失的捕获探针区域为纯合缺失:1)缺失的捕获探针区域的实际深度di小于等于深度阈值,其中,实际深度di=样本深度
×
肿瘤纯度c,样本深度为待测肿瘤样本的原始测序深度或归一化深度;2)缺失的捕获探针区域的dn不符合正态分布模型。
16.进一步地,深度归一化模块包括:原始深度统计模块,用于按照如下原则统计捕获区域的原始测序深度:a) read1和read2的重叠区域只统计一次,b) 发生插入缺失的reads纳入统计;样本内归一化模块,用于在样本内,以捕获探针区域为单位对原始测序深度进行第一次归一化处理,得到样本内归一化深度;样本间归一化模块,用于在样本间,对同一捕获探针区域的样本内归一化深度进行第二次归一化处理,得到同一捕获探针区域的归一化深度;标记模块,用于将待测肿瘤样本的和对照样本的归一化深度分别记为dn和pon;优选地,样本内归一化模块包括如下公式:每一捕获探针区域的原始测序深度/样本内所有捕获探针区域的原始测序深度的中位值;优选地,样本间归一化模块包括如下公式:同一捕获探针区域的样本内归一化深度/所有样本在同一捕获探针区域的样本内归一化深度的中位值。
17.进一步地,深度归一化模块还包括去除模块,用于去除原始测序深度为0的捕获探针区域;优选地,装置还包括:获取备选区域模块,用于将待测肿瘤样本中归一化后不满足正态分布模型的捕获探针区域作为备选分析区;优选地,纯合缺失推断模块用于当缺失的捕获探针区域位于备选分析区中时,推断存在缺失的捕获探针区域为纯合缺失。
18.进一步地,缺失区域初检模块包括:检测模块,用于以单个捕获探针区域为单位,检测待测肿瘤样本的归一化深度dn是否满足dn<待测肿瘤样本的归一化深度dn的均值
‑ꢀ
n*标准差,1.7≤n≤2.4,判断模块,用于在归一化深度dn满足dn<待测肿瘤样本的归一化深度dn的均值
‑ꢀ
n*标准差时,判定捕获探针区域存在缺失。
19.进一步地,纯合缺失推断模块包括肿瘤纯度c计算模块,肿瘤纯度c计算模块包括:杂合snp频率统计模块,用于选取对照样本中0.15≤af≤0.85的杂合snp,并从待测肿瘤样
本中找出相应杂合snp的突变频率;af矫正模块,用于以捕获探针区域为单位,从归一化处理后的测序数据中读取各杂合snp的支持read数,对对照样本中各杂合snp的af进行矫正,得到矫正后的af,其中,若双端测序的read 1和read 2都支持同一杂合snp,则只计算一次;方差异常区域筛查模块,用于以捕获探针区域为单位,分别统计对照样本和待测肿瘤样本的各个捕获探针区域的平均的归一化深度,并进一步分别计算对照样本和待测肿瘤样本的同一捕获探针区域内所有snp位点的归一化深度的方差,如果待测肿瘤样本的捕获探针区域内所有snp位点的归一化深度的方差超出对照样本在同一捕获探针区域的方差波动范围,则标记为异常区域;caf统计模块,用于在异常区域中,将待测肿瘤样本的杂合snp中相对于对照样本的杂合snp位点中基因频率下降的等位基因记为caf,若矫正后的af≥0.5,则caf=1
‑
矫正后的af;若矫正后的af<0.5,则caf=矫正后的af;肿瘤纯度c计算子模块,用于计算异常区域中,各捕获探针区域的log2(dn/pon)值,若捕获探针区域的log2(dn/pon)值<0,则表明捕获探针区域发生了杂合性缺失,此时肿瘤纯度c = (1
‑
2caf) / (1
‑
caf)。
20.进一步地,方差异常区域筛查模块进一步包括:区域合并模块,用于在计算各个捕获探针区域的平均的归一化深度时,如果相邻的捕获探针之间有重叠,则合并为一个捕获探针区域;和/或区域分割模块,用于在进行方差计算时,如果捕获探针区域所涵盖的物理距离大于等于1mb且待测肿瘤样本的方差超出对照样本在同一捕获探针区域的方差波动范围时,将捕获探针区域进一步分割成多个,然后再进行方差的计算。
21.进一步地,获取模块包括:质控模块,用于对待测肿瘤样本和对照样本的原始测序数据进行质控处理,得到有效数据;初比对模块,用于将有效数据与参考基因组序列进行比对,得到初步比对信息;重比对模块,用于对有效数据中含有indel位点的区域进行重新比对,得到比对结果数据;优选地,重比对模块包括:查找模块,用于根据已知的indel位点信息,查找有效数据中所有的indel区域,形成indel区域序列文件;重比对子模块,用于结合indel区域序列文件,对初步比对信息中的indel区域进行重新比对;优选地,质控模块包括:筛选模块,根据碱基测序质量、reads长度、n碱基含量对原始测序数据进行筛选;去除模块,用于同时去除原始测序数据中的引物序列和不能被引物序列识别的序列;优选地,待测肿瘤样本为前列腺癌石蜡包埋组织样本,原始测序数据是靶向捕获测序数据;更优选地,靶向捕获测序数据是靶向捕获如下14个同源重组修复基因的测序数据:brca1、brca2、atm、cdk12、palb2、brip1、rad54l、bard1、rad51b、rad51d、chek1、chek2、fancl及rad51c。
22.根据本技术的第三个方面,还提供了一种计算机可读存储介质,该存储介质包括存储的程序,其中,在程序运行时控制存储介质所在设备执行上述检测纯合缺失的方法。
23.根据本技术的第四个方面,还提供了一种处理器,该处理器用于运行程序,其中,程序运行时执行上述纯合缺失的方法。
24.应用本发明的技术方案,通过先利用正常对照样本的比对结果数据建立归一化深度pon,使不同捕获区域的原始测序深度转化为同一标准下的相对深度值,进而便于后续的分析。利用对照样本的归一化深度pon建立所有捕获探针在不同捕获区域的捕获效率的正态分布模型,利用该模型便于检测出待测肿瘤样本中存在不满足该模型分布规律的捕获探针区域。利用待测肿瘤样本的归一化深度dn,根据dn的异常,初步检测出某个区域是否存在缺失;然后通过考虑初步判定为缺失区域的实际深度是否低于深度阈值,或者是否满足捕获探针捕获效率的正态分布模型,来推断缺失区域是否为纯合缺失,该方法使得检测结果
更准确。
附图说明
25.构成本技术的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:图1示出了根据本发明的优选实施例中检测纯合缺失的方法的流程示意图;图2a至图2g分别示出了本技术实施例2中利用本技术的检测方法经各步骤处理后得到结果图。
具体实施方式
26.需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。下面详细描述本发明的实施例,具体描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。实施例中未注明具体技术条件者,按照相关领域内文献描述的技术条件或者相关产品说明书进行。所用软件或数据没有特殊说明者,均为可以通过网络获得的常规软件或数据。
27.术语解释:捕获探针区域:在本技术中大多数情况下与区域的含义相同,均指捕获探针所覆盖的区域。只有在某些特定情况下,比如相邻捕获探针之间的测序数据存在重叠时,将相邻的捕获探针区域进行合并而得到了一个覆盖范围更广的区域,此时,也将该覆盖范围更广的区域认为是一个捕获探针区域。
28.归一化(normallization):数据标准化的一种方式,将数据按比例缩放,使之落入一个小的特定区间。在比较和评价的指标处理中经常用到,去除数据单位的限制,将其转化为物量纲的纯数值,便于不同单位或量级的指标能够进行比较和加权。而归一化处理就是将数据统一映射到[0,1]区间上。
[0029]
在本技术一种典型的实施例中,提供了一种检测纯合缺失的方法,如图1所示,该方法包括:s1,获取待测肿瘤样本和对照样本的测序数据,并分别与参考基因组序列比对得到的比对结果数据,配对样本包括待测肿瘤样本和对照样本;s2,利用比对结果数据,分别统计待测肿瘤样本和对照样本的原始测序深度,对原始测序深度进行归一化处理,分别得到待测肿瘤样本的和对照样本的归一化深度,相应记为dn和pon;s3,利用对照样本的归一化深度pon建立所有捕获探针的正态分布模型;s4,利用待测肿瘤样本的归一化深度dn,初步检测待测肿瘤样本中存在缺失的捕获探针区域;s5,根据如下任意一种情况来判断存在缺失的捕获探针区域为纯合缺失:1)缺失的捕获探针区域的实际深度di小于等于深度阈值,其中,实际深度di=样本深度
×
肿瘤纯度c,样本深度为待测肿瘤样本的原始测序深度或归一化深度(两种深度均不影响结果的判定);2)缺失的捕获探针区域的dn不符合正态分布模型。
[0030]
本技术的检测纯合缺失突变的方法,通过先利用对照样本的比对结果数据建立归一化深度pon,使不同捕获区域的原始测序深度转化为同一标准下的相对深度值,进而便于后续的分析。利用对照样本的归一化深度pon建立所有捕获探针在不同捕获区域的捕获效率的正态分布模型,利用该模型便于检测出待测肿瘤样本中存在不满足该模型分布规律的捕获探针区域。利用待测肿瘤样本的归一化深度dn,根据dn的异常,初步检测出某个区域是否存在缺失;然后通过考虑初步判定为缺失区域的实际深度是否低于深度阈值,或者是否满足捕获探针捕获效率的正态分布模型,来推断缺失区域是否为纯合缺失,该方法使得检测结果更准确。
[0031]
需要说明的是,本技术的建立pon的对照样本是指与肿瘤样本相对的正常的非肿瘤样本,既可以取自与肿瘤样本同一个体的正常组织的样本,也可以是健康人的样本。此处优选采用健康人的正常样本来建立pon。此外,pon的建立与肿瘤样本的dn建立的时间顺序不限,但从逻辑上讲应该是先pon,之后再进行肿瘤样本的分析。
[0032]
本技术的上述方法中,基于对照样本的归一化深度pon构建所有捕获探针的正态分布模型的目的是获得捕获探针的捕获性能特征,以此检测待测肿瘤样本中的捕获探针的捕获性能是否与对照的特征相符合,若存在不符合的,则可以判断相应的捕获探针区域为异常区域,进一步根据该异常区域是否为初步判断的缺失的捕获探针区域,若是,则可以推断该缺失区域记为纯合缺失。
[0033]
上述方法中,纯合缺失的判断是基于肿瘤细胞具有异质性这一特点,从测序数据中通过计算出肿瘤纯度而换算得到缺失区域的具体深度即可判断是否为纯化缺失;而对于难以从测序数据中计算得到肿瘤纯度的情况,则通过对照样本建立捕获探针对不同区域的捕获特征,来检查待测肿瘤样本中同样的捕获特征的捕获特征是否与对照样本中相符,来进一步确认初步判定为缺失的区域属于纯合缺失。通过两者之一即可判定该方法进一步对现有方法中的缺失区域进行了纯合缺失判定,使得判定结果更精确。
[0034]
上述归一化处理的步骤按照现有高通量测序数据归一化处理的常规步骤进行处理即可。对于对照样本和待测肿瘤样本,归一化处理的步骤s2相同,均包括如下步骤:s21,按照如下原则统计捕获区域的所述原始测序深度:a) read1和read2的重叠区域只统计一次,b) 发生插入缺失的reads纳入统计;s22,在样本内,以捕获探针区域为单位对所述原始测序深度进行第一次归一化处理,得到样本内归一化深度;s23,在样本间,对同一捕获探针区域的样本内归一化深度进行第二次归一化处理,得到所述同一捕获探针区域的归一化深度;s24,将待测肿瘤样本的和对照样本的归一化深度分别记为dn和pon。
[0035]
具体地,在一种优选的实施例中,利用对照样本的比对结果数据,构建对照样本的归一化深度pon包括:按照如下原则统计捕获区域的原始测序深度:a) read1和read2的重叠区域只统计一次(目的在于去除测序造成的深度偏差),b) 发生插入缺失的reads纳入统计中(目的在于去除插入缺失对深度造成的偏差);在样本内(本技术中指一个panel为一个样本,而一个panel所涵盖的目的基因的数量,根据实际需要确定,比如本技术优选实施例中,一个panel涵盖了1460个目的基因)以捕获探针区域为单位对原始测序深度(此处指测序数据中直接统计获得的覆盖某一捕获探针区域的reads数,比如通过samtools depth统计得到的深度)进行第一次归一化处理,得到样本内归一化深度;在样本间,对同一捕获探针区域(比如样本a、b、c,每个样本中的同一个区域就是同一捕获探针区域)的样本内归一
化深度进行第二次归一化处理,得到对照样本在同一捕获探针区域归一化深度pon;优选地,第一次归一化处理通过每一捕获探针区域的原始测序深度/样本内所有捕获探针区域的原始测序深度的中位值得到;优选地,第二次归一化处理通过同一捕获探针区域的样本内归一化深度/所有样本在同一捕获探针区域的样本内归一化深度的中位值得到。
[0036]
上述样本内的归一化处理,以一个样本涵盖4000个基因为例,可以先统计每个基因的测序深度,然后计算这4000个基因的平均测序深度(比如,中位值),以每个基因的测序深度除以4000个基因的平均测序深度,获得每个基因的相对测序深度,作为该样本内每个基因的归一化深度。
[0037]
上述样本间的归一化处理,以200个样本为例,这200个样本中同一个a基因的样本内的归一化深度求的平均深度(比如,中位值),然后每个样本中的a基因的样本内归一化深度处于该中位值即可得到每个基因的归一化的pon。
[0038]
本技术上述对原始测序深度进行归一化方法,由于充分考虑了样本个体差异性、探针捕获误差、测序误差等系统误差,从而使得所统计的深度更接近样本实际深度。
[0039]
在一种优选的实施例中,利用待测肿瘤样本的比对结果数据及对照样本的归一化深度pon,统计待测肿瘤样本的归一化深度dn包括:按照如下原则统计捕获区域的原始测序深度:a) read1和read2的重叠区域只统计一次,b) 发生插入缺失的reads纳入统计中;c)在样本内以捕获探针区域为单位对原始测序深度进行第一次归一化处理,得到样本内归一化深度;以单个捕获探针区域为单位,通过同一捕获探针区域的样本内归一化深度/对照样本在同一捕获探针区域归一化深度pon进行第二次归一化处理,得到待测肿瘤样本在同一捕获探针区域的归一化深度dn。
[0040]
上述优选实施例中,待测样本的样本内的归一化数据之前的步骤与对照样本相同,对于某一捕获探针区域的深度通过以对照样本在该相同捕获探针区域的归一化深度pon为基准进行再次归一化,从而待测样本的深度统计更精准。
[0041]
为了避免测序未覆盖到的区域对检测结果造成假阳性的干扰,在一种优选的实施例中,在进行第一次归一化处理之前,该方法还包括:去除原始测序深度为0的区域(即单个探针捕获区域);优选地,在第一次归一化处理和/或第二次归一化处理后,该方法还包括将归一化后不满足正态分布的区域作为备选分析区。如果初步判断为缺失的区域同时还属于此处的备选分析区,则可以推断该缺失的捕获探针区域即是纯合缺失区域。
[0042]
上述基于对照样本的测序数据检测待测肿瘤样本中是否存在缺失的捕获探针区域的方法,可以采用现有的基于深度显著异常来进行初检。在一种具体的实施例中,缺失的捕获探针区域的初检步骤包括:以单个捕获探针区域为单位,检测待测肿瘤样本的归一化深度dn是否满足dn<待测肿瘤样本的归一化深度dn的均值
‑ꢀ
n*标准差,1.7≤n≤2.4,若是,判定捕获探针区域存在缺失。
[0043]
由于待测肿瘤样本中不可避免地带有一定比例的非肿瘤细胞,肿瘤细胞相对于非肿瘤细胞具有异质性,因而可以根据测序数据统计待测肿瘤样本中的肿瘤纯度,通过其纯度便于进一步精确计算来源与肿瘤细胞的突变的深度,为判定是否为纯合突变提供了更准确的判定依据。具体的肿瘤纯度的计算方法既可以采用现有方法进行计算,也可以在现有方法基础上进行改进得到。
[0044]
在一种优选的实施例中,肿瘤纯度c按照如下方法计算:
1)选取对照样本中0.15≤af≤0.85的杂合snp,从待测肿瘤样本中找出相应杂合snp的突变频率;2)以捕获探针区域为单位,从归一化处理后的测序数据中读取各杂合snp的支持read数,对对照样本中各杂合snp的af进行矫正,得到矫正后的af,其中,若双端测序的read 1和read 2都支持同一杂合snp,则只计算一次;3)以捕获探针区域为单位,分别统计对照样本和待测肿瘤样本的各个捕获探针区域的平均的归一化深度,并进一步分别计算对照样本和待测肿瘤样本的同一捕获探针区域内所有snp位点的归一化深度的方差,如果待测肿瘤样本的捕获探针区域内所有snp位点的归一化深度的方差超出对照样本在同一捕获探针区域的方差波动范围,则标记为异常区域;4)在异常区域中,将待测肿瘤样本的杂合snp中相对于对照样本的杂合snp位点中基因频率下降的等位基因记为caf,若矫正后的af≥0.5,则caf=1
‑
矫正后的af;若矫正后的af<0.5,则caf=矫正后的af;5)计算异常区域中,各捕获探针区域的log2(dn/pon)值,若捕获探针区域的log2(dn/pon)值<0,则表明捕获探针区域发生了杂合性缺失,此时肿瘤纯度c = (1
‑
2caf) / (1
‑
caf)。
[0045]
在某些优选的实施例中,在计算各个捕获探针区域的平均的归一化深度时,如果相邻的捕获探针之间有重叠,则合并为一个捕获探针区域;优选地,在进行方差计算时,如果捕获探针区域所涵盖的物理距离大于等于1mb且待测肿瘤样本的方差超出对照样本在同一捕获探针区域的方差波动范围的,则将捕获探针区域进一步分割成多个,然后再进行方差的计算。
[0046]
通过利用杂合snp位点的矫正后的等位基因频率以及不同捕获探针分区内的平均的均一化深度的方差与对照样本之间的差异,筛选出存在异常的区域,根据肿瘤样本的异常区域内存在杂合性缺失的snp位点来推算肿瘤纯度。
[0047]
在一种优选的实施例中,获取待测肿瘤样本和对照样本的测序数据,并分别与参考基因组序列比对得到的比对结果数据包括:对待测肿瘤样本和对照样本的原始测序数据进行质控处理,得到有效数据;将有效数据与参考基因组序列进行比对(比如,利用bwa软件进行比对),得到初步比对信息;对有效数据中含有indel位点的区域进行重新比对(比如,利用gatk4软件进行比对),得到比对结果数据;优选地,重新比对包括:根据已知的indel位点信息(比如参考已知的人类基因组参考信息以及1000genomic中的indel位点),查找有效数据中所有的indel区域,形成indel区域序列文件;结合indel区域序列文件,对初步比对信息中的indel区域进行重新比对;优选地,质控处理包括: 根据碱基测序质量、reads长度、n碱基含量对原始测序数据进行筛选,同时去除原始测序数据中的引物序列和不能被引物序列识别的序列。
[0048]
测序下机的原始数据(raw data)通常需要经过去除低质量的测序数据等预处理才能获得有效数据(又叫clean data)。
[0049]
根据碱基测序质量、reads长度、n碱基含量对原始测序数据进行筛选筛选和去除,避免测序过程中的低质量数据对后续数据分析造成的干扰,提高后续分析结果的准确性。具体的去除碱基质量小于20的碱基占比超过40%的reads;去除n碱基大于1个的reads;去除
长度小于100bp的reads。
[0050]
在进行目标区域的靶向测序时,每条测序read均通过测序引物测序得到,因而获得的原始测序数据中每条read的两端均包含测序引物序列。在前处理过程中,根据已知的测序引物信息将每条read中的测序引物序列去除,以提高后续分析效率。
[0051]
上述前处理步骤中,利用测序引物序列去除低质量数据后的测序数据中序列进行识别,得到能够识别上的含测序引物序列的reads和不能识别对应测序引物的reads,从而去除掉能识别上的序列中对应的测序引物序列,同时去除不能识别对应测序引物的reads,得到处理合格的,符合分析输入需求的双端测序fastq序列。
[0052]
上述与参考基因组序列进行比对,得到比对结果的比对步骤采用现有的比对方法进行比对即可。本技术优选的实施例中,该比对步骤包括:将有效数据与参考基因组序列进行全局比对,得到初步比对信息;对初步比对信息中的indel区域进行重新比对,得到比对结果。优选地,重新比对包括:根据已知的indel位点信息,查找合并后数据中所有的indel区域,形成indel区域序列文件;结合indel区域序列文件,对初步比对信息中的indel区域进行重新比对。
[0053]
上述已知的indel位点信息可以是已有的人类参考基因组序列和千人基因组计划推出的indel位点信息,或者其他方式确定的已知的indel位点信息。经过两次比对后的比对结果更准确。
[0054]
实施例2本实施例中检测肿瘤纯合缺失的简要步骤如下:1)测序数据质控:去除掉低质量的测序序列,包含测序接头的reads。
[0055]
2)序列比对:将得到的测序合格序列进行人类全基因组序列比对,生成bam文件。
[0056]
3)比对结果矫正:对2)比对的结果,进一步进行矫正分析(如indel区域重比对),以得到每条测序reads在基因组上的准确定位信息,以便后续分析。
[0057]
4)计算基因组的覆盖深度。
[0058]
5)正常样本(指对照样本,比如癌旁组织)模型构建。
[0059]
6)检测样本纯合缺失分析。
[0060]
详细检测流程如下:1.测序数据的预处理根据碱基测序质量、reads长度、n碱基含量对原始测序数据进行筛选,去除测序reads中的接头序列,避免这些因素对后续数据分析造成的干扰,提高后续分析结果的准确性。
[0061]
2. 序列比对将处理得到的合格序列比对到人类参考基因组上,得到测序数据的初步比对结果。
[0062]
3. 比对结果矫正若序列中存在碱基的插入或缺失变异时,会对比对结果的正确性产生影响,并会直接影响周边变异的检测性。因而,对上一步得到的比对初步结果,进一步处理,对indel区域进行重新比对,以得到每条测序reads在基因组上的准确定位信息。
[0063]
具体来说,就是利用已有的人类参考基因组序列和千人基因组计划推出的indel
位点信息,找到待测肿瘤样本中所有需要进行重新比对的区域,形成interval文件;然后结合该interval文件,对初步比对结果中的indel变异类型进行二次比对。以此得到最终的比对结果bam文件。
[0064]
4. 统计测序深度对上一步比对得到的bam文件进行深度统计,特别的以碱基为单位统计深度的过程中,read1和read2的overlap区域应当只统计一次,发生插入缺失的reads应当纳入深度统计中。此时得到的是原始测序深度,其中,待测肿瘤样本的原始测序深度如图2a所示。
[0065]
5. 分析样本的变异分析样本的突变,并计算突变的频率。配对样本中的肿瘤样本中,去掉配对正常样本中的突变,得到肿瘤突变频率。
[0066]
6. 构建正常样本模型 (pon,即panel of normal)1)根据上一步得到的正常样本panel捕获探针区域内每一个碱基位点的深度数据,去除深度为0的位点,得到深度dab,其中a代表不同的样本,b代表不同的位点。d34代表第3个样本第4个位点的深度。
[0067]
2)样本内深度归一化,计算每个样本的平均(median )深度mai = median([da1,da2...dan],n∈b)。对每个样本内的位点深度进行归一化dnab = dab/mai。
[0068]
3)在样本内深度归一化的基础上进行样本间深度归一化,对同一个位点不同样本间,使用样本内归一化后的深度dnab,计算median深度mmnb = median([dn1b,dn2b...dnnb],n∈a)。对dnnb进行样本间归一化得到dnnnb = dnab/mmib,dnnnb服从正态分布x
∼
n(μ,σ),计算得到μ和σ。
[0069]
7. 处理检测样本深度数据1)深度为0区域作为备选分析区域并从深度数据中去除,保留的区域进行样本内归一化dn = d/median(depth),按照对照的正常样本的pon建立的正态分布模型,对待测样本的dn进行分正态性检验,不符合的区域作为备选分析区域并标记;2)对于不同区域的dn进行样本间归一化,dnn = dn / mmnb(pon构建是计算的median深度),检验dnn是否符合上述正态分布x
∼
n(μ,σ),不符合的作为备选分析区域进行标记。即根据归一化深度初步检测得到的存在缺失的捕获探针区域。
[0070]
图2b示出了待测肿瘤样本的归一化处理后在不同捕获探针区域(竖线所示分区即为一个捕获探针区域)内的平均深度(横线所示)、各捕获探针区域内杂合snp位点的基因频率(数字所示)。
[0071]
8. 肿瘤纯度鉴定1)选取对照样本中突变中的杂合snp位点,0.15 <= af <= 0.85,从待测肿瘤样本中找出这些杂合snp位点的突变频率。
[0072]
2)计算矫正后的af:从samtools mpileup中读取snp的支持reads,如果r1和r2(即一条read名字出现两次)都支持snp,只计算一次。
[0073]
其中,对照样本中各杂合snp位点的af分布如图2c所示,待测肿瘤样本各杂合snp位点的af分布在矫正前和矫正后分别如图2d和图2e所示。图2f上显示的是待测肿瘤样本的各捕获探针区域(各竖线间隔的区域)内杂合snp位点矫正后的基因频率(数字所示)。
[0074]
3)以捕获探针为单位,计算一个捕获探针区域的平均深度。如果相邻探针之间有
重叠则合并为一个区域,计算同一个区域内所有位点深度的方差,如果方差在对照正常样本同一区域方差波动范围内,则为正常,反之标记为异常区域。(如果方差大于0.4的区域大于一定的物理距离,比如大于1mb或包含多个基因时,可以再细分成小区域后,再进行方差计算(以对照正常样本的每个捕获区域的平均深度的方差分布模型为参照进行检测,其体现的是每个捕获探针的捕获性能特征,如果待测肿瘤样本中每个捕获探针区域的平均深度与对照样本的方差分布模型不符合,则相应的捕获探针区域即为异常区域。异常区域包括缺失区域或扩增区域)。
[0075]
4)计算log2(dn/pon),定义caf为肿瘤样本杂合位点的2个allele位点中相对于配对的正常样本的位点频率下降的alllele。例如:正常样本中a:t=0.6:0.4,肿瘤样本中a:t=0.7:0.3,则caf=0.3。
[0076]
5)如果肿瘤样本在某个捕获探针区域的log2<0,则该捕获探针区域发生了杂合性缺失,此时肿瘤纯度c = (1
‑
2caf) / (1
‑
caf)。相应地,缺失区域的实际深度d1=样本深度*肿瘤纯度c。
[0077]
6)如果肿瘤样本在某个捕获探针区域的log2>0,则该捕获探针区域的某一个单体型发生了扩增,则肿瘤纯度c = (1
‑
2caf) / (caf*(t
‑
2)) 其中t为总拷贝数。
[0078]
9.纯合缺失判定1)综合考虑如下情况,如果某个捕获探针区域符合以下条件中的2条,则判断此捕获探针区域为纯合缺失区域。
[0079]
a、缺失的捕获探针区域的实际深度di小于等于深度阈值,其中,实际深度di=样本深度
×
肿瘤纯度c,样本深度为待测肿瘤样本的原始测序深度或归一化深度;b、缺失的捕获探针区域的dn不符合正态分布模型。
[0080]
具体可视化的判定结果如图2g所示。图2g中,以竖线间隔的各个捕获探针区域为单位,最上面的数字代表的是该区域的拷贝数,正常拷贝数为2,大于2的表示相应捕获探针区域存在扩增,小于2的区域表示相应捕获探针区域存在缺失。而1表示杂合性缺失,0表示纯合缺失。a或b表示基因型,基因型下面的一行的数字代表相应区域的肿瘤纯度,每个区域的值不一致是因为肿瘤异质性(肿瘤细胞的亚克隆)导致的。一般会取最高的一个值代表整个样本的肿瘤纯度。
[0081]
需要说明的是,如果某一个区域的固有特征会导致此区域的测序深度为0,那么在建立pon时这一特征会被记录,认为深度为0是正常状态。反之,如果建立的pon没有记录此特征,则在某区域的测序深度为0时认为发生了纯合缺失。
[0082] 实施例3 基于二代测序数据进行纯合缺失检测1. pon构建a) 选取4013例正常血液样本,使用14个同源重组修复基因panel(brca1、brca2、atm、cdk12、palb2、 brip1、rad54l、bard1、rad51b、rad51d、chek1、chek2、fancl及rad51c)进行捕获并用illumina平台进行测序。
[0083]
b) 测序下机数据使用fastp软件进行质控,生成clean data。
[0084]
c)使用bwa软件将clean data比对到hg19参考基因组。
[0085]
d)使用gatk4对bam文件进行重比对。
[0086]
e)使用samtools depth统计捕获区域的深度,read1和read2的overlap区域只统
计一次,发生插入缺失的reads计入深度统计中。
[0087]
f)计算样本深度的中位值median(样本深度),计算探针连续区间的median深度median(区域深度);g) 进行样本内深度归一化,归一化的深度=median(区域深度)/median(样本深度)h) 在上一步的基础上,以panel区域为单位,对4013个样本间的同一区域进行归一化,计算此区域深度的中值median(归一化深度),按区域归一化的深度=归一化深度/median(归一化深度)。这个区域的4013个深度服从正态分布,计算出μ,σ。
[0088]
2.检测样本基础分析a)选取20对肿瘤
‑
血液配对样本,捕获、测序、质控、比对、深度统计和样本内归一化的处理步骤与上述pon构建方法中对应步骤相同。
[0089]
b)以捕获探针区域为单位,分别除以pon中的median(归一化深度)进行第二次归一化,得到最终的归一化深度dn,如果dn < dn的均值
ꢀ‑ꢀ
2*标准差,即认为此捕获探针区域可能是缺失。
[0090]
c)同实施例2中第8)步,通过杂合snp位点在配对样本的正常(normal)中和肿瘤(tumor)中的变化,来鉴定肿瘤纯度c。
[0091]
d) 纯合缺失实际深度di = 样本深度*肿瘤纯度c,来进一步推算上述存在缺失的捕获探针区域是否为纯合缺失。若di小于深度阈值,则推断为纯合缺失。此实施例中,深度阈值为1/4的样本深度,即di小于1/4的样本深度时,认为是纯合缺失。
[0092]
e)如果难以获得肿瘤纯度c,则根据肿瘤样本中各捕获探针区域的dn分布是否满足实施例2中对照样本的归一化深度pon建立的正态分布模型,若不符合,则同样能够推算上述存在缺失的捕获探针为纯合缺失。
[0093]
3.分析结果需要说明的是,对于前述的各方法实施例,为了简单描述,故将其都表述为一系列的动作组合,但是本领域技术人员应该知悉,本发明并不受所描述的动作顺序的限制,因为依据本发明,某些步骤可以采用其他顺序或者同时进行。其次,本领域技术人员也应该知悉,说明书中所描述的实施例均属于优选实施例,所涉及的动作并不一定是本发明所必须的。
[0094]
通过以上的实施方式的描述可知,本领域的技术人员可以清楚地了解到本技术可借助软件加必需的检测仪器等硬件设备的方式来实现。基于这样的理解,本技术的技术方案中数据处理的部分可以以软件产品的形式体现出来,该计算机软件产品可以存储在存储介质中,如rom/ram、磁碟、光盘等,包括若干指令用以使得一台计算机设备(可以是个人计算机,服务器,或者网络设备等)执行本技术各个实施例或者实施例的某些部分的方法。
[0095]
以下部分为能够执行上述微卫星状态的检测方法的仪器设备实施例4本实施例提供了一种检测纯合突变的装置,该装置包括:获取模块、深度归一化模块、探针捕获特征模型构建模块、缺失区域初检模块及纯合缺失推断模块,其中,获取模块,用于获取待测肿瘤样本和对照样本的测序数据,并分别与参考基因组序列比对得到的比对结果数据,配对样本包括待测肿瘤样本和对照样本;
深度归一化模块,用于利用比对结果数据,分别统计待测肿瘤样本和对照样本的原始测序深度,对原始测序深度进行归一化处理,分别得到待测肿瘤样本的和对照样本的归一化深度,相应记为dn和pon;探针捕获特征模型构建模块:用于利用对照样本的归一化深度pon建立所有捕获探针的正态分布模型;缺失区域初检模块,用于利用待测肿瘤样本的归一化深度dn,初步检测待测肿瘤样本中存在缺失的捕获探针区域;纯合缺失推断模块,用于根据如下任意一种情况来推断存在缺失的捕获探针区域为纯合缺失:1)缺失的捕获探针区域的实际深度di小于等于深度阈值,其中,实际深度di=样本深度
×
肿瘤纯度c,样本深度为待测肿瘤样本的原始测序深度或归一化深度;2)缺失的捕获探针区域的dn不符合正态分布模型。
[0096]
上述装置,通过先利用深度归一化模块根据对照样本的比对结果数据建立归一化深度pon,使不同捕获区域的原始测序深度转化为同一标准下的相对深度值,进而便于后续的分析。然后执行探针捕获特征模型构建模块,利用对照样本的归一化深度pon建立所有捕获探针在不同捕获区域的捕获效率的正态分布模型,利用该模型便于检测出待测肿瘤样本中存在不满足该模型分布规律的捕获探针区域。通过执行缺失区域初检模块利用待测肿瘤样本的归一化深度dn,根据dn的异常,初步检测出某个区域是否存在缺失;然后执行纯合缺失推断模块,通过考虑初步判定为缺失区域的实际深度是否低于深度阈值,或者是否满足捕获探针捕获效率的正态分布模型,来推断缺失区域是否为纯合缺失,该装置使得检测结果更准确。
[0097]
可选地,深度归一化模块包括:原始深度统计模块,用于按照如下原则统计捕获区域的原始测序深度:a) read1和read2的重叠区域只统计一次,b) 发生插入缺失的reads纳入统计;样本内归一化模块,用于在样本内,以捕获探针区域为单位对原始测序深度进行第一次归一化处理,得到样本内归一化深度;样本间归一化模块,用于在样本间,对同一捕获探针区域的样本内归一化深度进行第二次归一化处理,得到同一捕获探针区域的归一化深度;标记模块,用于将待测肿瘤样本的和对照样本的归一化深度分别记为dn和pon;优选地,样本内归一化模块包括如下公式:每一捕获探针区域的原始测序深度/样本内所有捕获探针区域的原始测序深度的中位值;优选地,样本间归一化模块包括如下公式:同一捕获探针区域的样本内归一化深度/所有样本在同一捕获探针区域的样本内归一化深度的中位值。
[0098]
可选地,深度归一化模块还包括去除模块,用于去除原始测序深度为0的捕获探针区域;优选地,装置还包括:获取备选区域模块,用于将待测肿瘤样本中归一化后不满足正态分布模型的捕获探针区域作为备选分析区;优选地,纯合缺失推断模块用于当缺失的捕获探针区域位于备选分析区中时,推断存在缺失的捕获探针区域为纯合缺失。
[0099]
可选地,缺失区域初检模块包括:检测模块,用于以单个捕获探针区域为单位,检测待测肿瘤样本的归一化深度dn是否满足dn<待测肿瘤样本的归一化深度dn的均值
‑ꢀ
n*标准差,1.7≤n≤2.4;判断模块,用于在归一化深度dn满足dn<待测肿瘤样本的归一化深度dn的均值
‑ꢀ
n*标准差时,判定捕获探针区域存在缺失。
[0100]
可选地,纯合缺失推断模块包括肿瘤纯度c计算模块,肿瘤纯度c计算模块包括:杂
合snp频率统计模块,用于选取对照样本中0.15≤af≤0.85的杂合snp,并从待测肿瘤样本中找出相应杂合snp的突变频率;af矫正模块,用于以捕获探针区域为单位,从归一化处理后的测序数据中读取各杂合snp的支持read数,对对照样本中各杂合snp的af进行矫正,得到矫正后的af,其中,若双端测序的read 1和read 2都支持同一杂合snp,则只计算一次;方差异常区域筛查模块,用于以捕获探针区域为单位,分别统计对照样本和待测肿瘤样本的各个捕获探针区域的平均的归一化深度,并进一步分别计算对照样本和待测肿瘤样本的同一捕获探针区域内所有snp位点的归一化深度的方差,如果待测肿瘤样本的捕获探针区域内所有snp位点的归一化深度的方差超出对照样本在同一捕获探针区域的方差波动范围,则标记为异常区域;caf统计模块,用于在异常区域中,将待测肿瘤样本的杂合snp中相对于对照样本的杂合snp位点中基因频率下降的等位基因记为caf,若矫正后的af≥0.5,则caf=1
‑
矫正后的af;若矫正后的af<0.5,则caf=矫正后的af;肿瘤纯度c计算子模块,用于计算异常区域中,各捕获探针区域的log2(dn/pon)值,若捕获探针区域的log2(dn/pon)值<0,则表明捕获探针区域发生了杂合性缺失,此时肿瘤纯度c = (1
‑
2caf) / (1
‑
caf)。
[0101]
可选地,方差异常区域筛查模块进一步包括:区域合并模块,用于在计算各个捕获探针区域的平均的归一化深度时,如果相邻的捕获探针之间有重叠,则合并为一个捕获探针区域;和/或区域分割模块,用于在进行方差计算时,如果捕获探针区域所涵盖的物理距离大于等于1mb且待测肿瘤样本的方差超出对照样本在同一捕获探针区域的方差波动范围时,将捕获探针区域进一步分割成多个,然后再进行方差的计算。
[0102]
可选地,获取模块包括:质控模块,用于对待测肿瘤样本和对照样本的原始测序数据进行质控处理,得到有效数据;初比对模块,用于将有效数据与参考基因组序列进行比对,得到初步比对信息;重比对模块,用于对有效数据中含有indel位点的区域进行重新比对,得到比对结果数据;优选地,重比对模块包括:查找模块,用于根据已知的 indel位点信息,查找有效数据中所有的indel区域,形成indel区域序列文件;重比对子模块,用于结合indel区域序列文件,对初步比对信息中的indel区域进行重新比对;优选地,质控模块包括:筛选模块,根据碱基测序质量、reads长度、n碱基含量对原始测序数据进行筛选;去除模块,用于同时去除原始测序数据中的引物序列和不能被引物序列识别的序列;优选地,待测肿瘤样本为前列腺癌石蜡包埋组织样本,原始测序数据是靶向捕获测序数据;更优选地,靶向捕获测序数据是靶向捕获如下14个同源重组修复基因的测序数据:brca1、brca2、atm、cdk12、palb2、 brip1、rad54l、bard1、rad51b、rad51d、chek1、chek2、fancl及rad51c。
[0103]
实施例5本实施例提供了一种存储介质,该存储介质包括存储的程序,其中,在程序运行时控制存储介质所在设备执行上述任一种检测纯合突变的方法。
[0104]
本实施例还提供了一种处理器,处理器用于运行程序,其中,程序运行时执行上述任一种检测纯合突变的方法。
[0105]
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:通过利用对照样本的比对结果数据建立归一化深度pon,建立了捕获探针在不同捕获区域的捕获效率的性能特征,体现出每个探针捕获的性能差异,进而检测待测肿瘤样本的各捕获探针区域的归一化深度dn,筛选得到不满足探针捕获性能特征的区域,进而能够进一步确认缺失
的区域为纯合缺失,这样的判定方法能够使否存在缺失区域的判定结果相对更准确;另外通过综合考虑肿瘤纯度及缺失的实际深度来确认某一区域的缺失是否为纯合缺失,也有利于提高缺失判定的精度。该方法通过建立pon来记录固有特征导致的深度为0的区域并视为测序未覆盖到的,若未被pon记录的深度为0的区域,则认为所检测到的缺失是真实的纯合,而非测序未覆盖到,进而使得检测结果更准确。
[0106]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1