二倍半萜类化合物的制备方法及其在抗临床耐药细菌方面的应用
1.本发明属于抗菌活性研究技术领域,具体涉及一种二倍半萜类化合物quiannulatic acid的制备方法及其在抗临床耐药细菌方面的应用。
背景技术:
2.近年来,随着抗生素类药物在临床上的不合理使用,细菌耐药性问题越来越严峻,甚至催生了“超级细菌”,即具有多重耐药性的细菌,如1961年英国je vons首次发现了耐甲氧西林金黄色葡萄球菌(mrsa)的存在,1992年到200 3年间mrsa在金黄色葡萄球菌中的比重由35.9%增加至64.4%。mrsa不仅对甲氧西林和大多数β-内酰胺类抗生素,对其他抗生素如氯霉素、克林霉素、四环素、氨基糖胺类等药物也有耐药性,根据全国细菌耐药监测网显示mrsa 对大部分抗菌药耐药率在60%以上。此外,耐万古霉素肠球菌(vre),自从1 980s首次报道以来,在全球范围很快蔓延,由于其对多种抗生素耐药,vre已成为治疗肠球菌感染的难题。因此,寻找针对mrsa和vre治疗药物成为当务之急。
3.面对越来越多耐药细菌的出现,抗生素研发却从80年代“抗生素黄金时期”后逐渐滞后,这样的局面对人类的健康和安全造成了极大威胁,解决细菌耐药问题刻不容缓,目前也引起了全球的广泛关注。因此,为了能够有效地防治和治疗细菌感染疾病,解决目前抗生素药物严重不足问题,发现和开发新型高效、低毒抗细菌药物已成为当今国际医药研究领域的重点之一。
4.微生物天然产物具有结构新颖、活性独特等特点,如青霉素、链霉素、红霉素等都是来源于微生物,微生物天然产物长期以来在药物发现和开发中占据着非常重要的地位。1981-2019全世界所批准上市的小分子实体药物数据表明,在过去近40年所批准上市的抗感染药物共401个,其中抗细菌药物有162个,而在162个抗细菌药物中,其中直接或间接源于天然产物的有90个,约占56%,说明天然产物在抗感染治疗,尤其是在抗细菌药物中占有率很高。
5.植物内生真菌由于其生存环境特殊,在与宿主植物长期复杂的共生关系中协同进化,使其具有独特的代谢体系,不仅可以产生结构新颖、活性多样的次级代谢产物,有些还可以产生与宿主植物相似的生物活性物质。大量研究表明,大多数植物内生真菌次级代谢产物具有显著抗细菌活性,并从中分离得到许多抗细菌活性单体化合物,因此,从植物内生真菌中寻找新型抗细菌药物或先导化合物有非常大的研究空间。
技术实现要素:
6.本发明的主要目的在于提供一种二倍半萜类化合物quiannulatic acid的制备方法及其在抗临床耐药细菌方面的应用。所述二倍半萜类化合物quiannulati c acid在低浓度的情况下可有效抑制2株临床耐药细菌生长,具有非常大的应用潜能。
7.为实现上述目的,本发明采用的技术方案如下:
8.二倍半萜类化合物作为制备抗临床耐药细菌药剂的应用,所述的二倍半萜类化合物结构式如式(ⅰ)所示;
[0009][0010]
进一步,优选的是,所述临床耐药细菌为atcc 43300耐甲氧西林金黄色葡萄球菌和atcc 51299万古霉素耐药粪肠球菌。
[0011]
进一步,优选的是,所述的二倍半萜类化合物的制备方法包括如下步骤:
[0012]
步骤(1),将真菌emericella sp.xl029使用大米固体培养基进行培养,静置培养28-30天,得到发酵产物;
[0013]
步骤(2),用甲醇萃取步骤(1)得到的发酵产物,将所得到的萃取液减压浓缩得到粗浸膏;
[0014]
步骤(3),将步骤(2)得到的粗浸膏用水溶解后,用乙酸乙酯反萃,取乙酸乙酯相减压浓缩,得到浸膏;
[0015]
步骤(4),将步骤(3)得到的浸膏干法上样至正相硅胶柱,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到 7个组分a
–
g;
[0016]
步骤(5),将步骤(4)得到的组分b干法上样至正相硅胶柱层析,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到5个亚组分b1
–
b5;
[0017]
步骤(6),对步骤(5)得到的亚组分b1进行纯化,得到式(ⅰ)所示的二倍半萜类化合物。
[0018]
进一步,优选的是,步骤(2)中,所述的萃取方式是加入等体积甲醇在室温条件下超声0.9-1.1h,然后室温条件下静置9-11h,取上清,减压浓缩;萃取次数为2-4次。
[0019]
进一步,优选的是,步骤(3)中,所用的水和乙酸乙酯的体积相同。
[0020]
进一步,优选的是,步骤(4)中,浸膏干法上样的具体方法为:将浸膏用 10倍体积甲醇溶解后,用浸膏等质量80-100目硅胶吸附拌样后上样;
[0021]
所述的正相硅胶柱中装柱硅胶为100-200目的正相硅胶;
[0022]
梯度洗脱采用的流动相按洗脱顺序依次为石油醚、二氯甲烷和甲醇的混合溶剂、甲醇,二氯甲烷和甲醇的混合溶剂中二氯甲烷和甲醇的体积比依次为10 0:1、80:1、50:1、20:1、10:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到a
–
g共7个组分。
[0023]
进一步,优选的是,步骤(5)中,浸膏干法上样的具体方法为:将组分b 用10倍体积甲醇溶解后,用组分b等质量80-100目硅胶吸附拌样后上样;
[0024]
所述的正相硅胶柱中装柱硅胶为100-200目的正相硅胶;
[0025]
梯度洗脱采用的流动相为石油醚和乙酸乙酯,石油醚和乙酸乙酯的体积比依次为100:1、60:1、30:1、10:1、1:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到b1
–
b5共5个亚组分。
[0026]
进一步,优选的是,步骤(6)中,所述的纯化先采用经凝胶色谱纯化,再采用制备液
相(hplc)纯化。
[0027]
进一步,优选的是,步骤(6)中,凝胶色谱纯化采用的凝胶柱为sephad ex lh-20,以体积比为1:1的二氯甲烷和甲醇的混合溶剂作为流动相,然后进行hplc;
[0028]
凝胶色谱纯化时,将亚组分b1用2倍体积甲醇溶解后上样,经tlc监测,合并相同的部分。
[0029]
所述的hplc色谱条件为流动相为体积比为19:1甲醇和水的混合溶剂,流速为1ml/min,收集开始出峰时间为5.6min的组分,采用安捷伦c
18
反向色谱柱,规格为:4.6mmx250nm,5um;柱温:30℃,每次进样量:20ul,采用紫外检测器,检测波长为210nm。
[0030]
本发明梯度洗脱时,采用tlc监测,合并相同部分。
[0031]
本发明同时提供一种抗临床耐药细菌药剂,所述的活性成分中含有二倍半萜类化合物,所述的二倍半萜类化合物结构式如式(ⅰ)所示;
[0032][0033]
本发明所述临床耐药细菌为耐甲氧西林金黄色葡萄球菌atcc 43300(me thicillin-resistant staphylcoccus aureus)和万古霉素耐药粪肠球菌atcc 51 299(vancomycin-resistant enterococcus faecalis)。该标准菌株购进于广东省微生物菌种保藏中心(gdmcc),菌株编号为gdmcc 1.1263=atcc 4330 0、gdmcc 1.1332=atcc 51299。
[0034]
所述的二倍半萜类化合物对耐甲氧西林金黄色葡萄球菌和万古霉素耐药粪肠球菌这2株临床耐药细菌的最小抑菌浓度mic均为2μg/ml。
[0035]
本发明与现有技术相比,其有益效果为:
[0036]
(1)本发明提供了一个二倍半萜类化合物quiannulatic acid在2株临床耐药细菌的应用,所述的二倍半萜类化合物能够在低浓度的情况下,抑制2株临床耐药细菌的生长;化合物对耐甲氧西林金黄色葡萄球菌和万古霉素耐药粪肠球菌这2株临床耐药细菌的最小抑菌浓度mic均为2μg/ml。由此可见,所述的二倍半萜类化合物在临床耐药细菌上具有非常大的应用潜能。
[0037]
(2)本发明还提供了该化合物的制备方法,能够快速准确高效的从真菌e mericella sp.xl029中制备获得所述的二倍半萜类化合物。
附图说明
[0038]
图1为本发明所述的二倍半萜类化合物的化学结构式;
[0039]
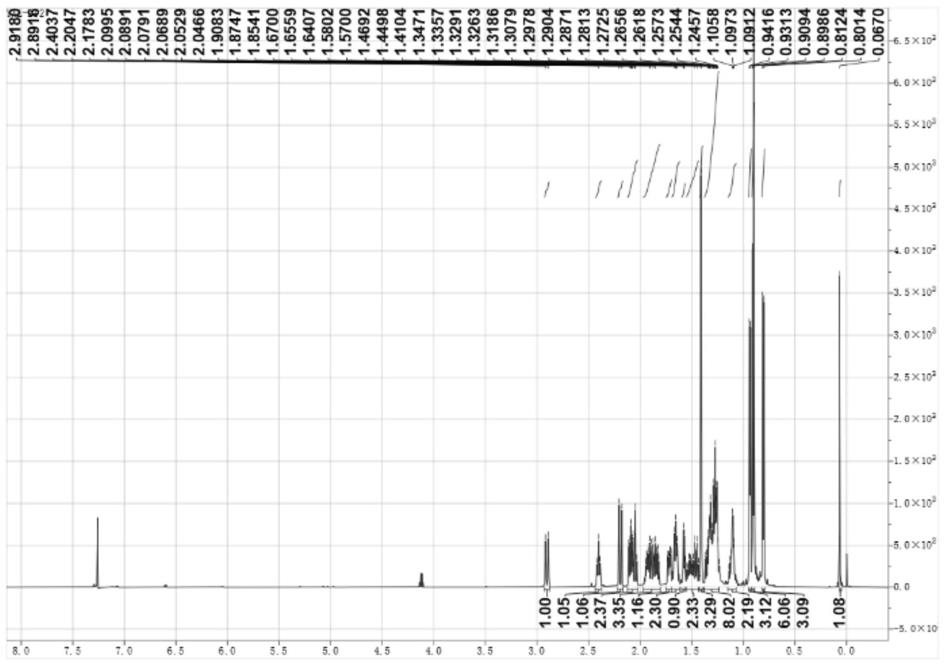
图2为本发明所述的二倍半萜类化合物的1h nmr图;
[0040]
图3为本发明所述的二倍半萜类化合物的
13
c nmr图。
[0041]
真菌emericella sp.xl029,已于2021年9月9日保藏于中国典型培养物保藏中心,保藏编号cctcc m 20211154,保藏地址为湖北省武汉市武昌区八一路299号,武汉大学中国典型培养物保藏中心。
具体实施方式
[0042]
下面结合实施例对本发明作进一步的详细描述。
[0043]
本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用材料或设备未注明生产厂商者,均为可以通过购买获得的常规产品。
[0044]
本发明所述的大米固体培养基的配方为:大米和水按质量1:1混合后,于 121℃下灭菌25min,即得。
[0045]
实施例1
[0046]
二倍半萜类化合物作为制备抗临床耐药细菌药剂的应用,所述的二倍半萜类化合物结构式如式(ⅰ)所示;
[0047][0048]
所述临床耐药细菌为atcc 43300耐甲氧西林金黄色葡萄球菌和atcc 5 1299万古霉素耐药粪肠球菌。
[0049]
实施例2
[0050]
二倍半萜类化合物的制备方法包括如下步骤:
[0051]
步骤(1),将真菌emericella sp.xl029使用大米固体培养基进行培养,静置培养29天,得到发酵产物;
[0052]
步骤(2),用甲醇萃取步骤(1)得到的发酵产物,将所得到的萃取液减压浓缩得到粗浸膏;
[0053]
步骤(3),将步骤(2)得到的粗浸膏用水溶解后,用乙酸乙酯反萃,取乙酸乙酯相减压浓缩,得到浸膏;
[0054]
步骤(4),将步骤(3)得到的浸膏干法上样至正相硅胶柱,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到 7个组分a
–
g;
[0055]
步骤(5),将步骤(4)得到的组分b干法上样至正相硅胶柱层析,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到5个亚组分b1
–
b5;
[0056]
步骤(6),对步骤(5)得到的亚组分b1进行纯化,得到式(ⅰ)所示的二倍半萜类化合物。
[0057]
实施例3
[0058]
二倍半萜类化合物的制备方法包括如下步骤:
[0059]
步骤(1),将真菌emericella sp.xl029使用大米固体培养基进行培养,静置培养28天,得到发酵产物;
[0060]
步骤(2),用甲醇萃取步骤(1)得到的发酵产物,将所得到的萃取液减压浓缩得到粗浸膏;
[0061]
步骤(3),将步骤(2)得到的粗浸膏用水溶解后,用乙酸乙酯反萃,取乙酸乙酯相减压浓缩,得到浸膏;
[0062]
步骤(4),将步骤(3)得到的浸膏干法上样至正相硅胶柱,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到 7个组分a
–
g;
[0063]
步骤(5),将步骤(4)得到的组分b干法上样至正相硅胶柱层析,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到5个亚组分b1
–
b5;
[0064]
步骤(6),对步骤(5)得到的亚组分b1进行纯化,得到式(ⅰ)所示的二倍半萜类化合物。
[0065]
步骤(2)中,所述的萃取方式是加入等体积甲醇在室温条件下超声0.9h,然后室温条件下静置9h,取上清,减压浓缩;萃取次数为2次。
[0066]
步骤(3)中,所用的水和乙酸乙酯的体积相同。
[0067]
步骤(4)中,浸膏干法上样的具体方法为:将浸膏用10倍体积甲醇溶解后,用浸膏等质量80目硅胶吸附拌样后上样;
[0068]
所述的正相硅胶柱中装柱硅胶为100目的正相硅胶;
[0069]
梯度洗脱采用的流动相按洗脱顺序依次为石油醚、石油醚和乙酸乙酯的混合溶剂、甲醇,石油醚和乙酸乙酯的混合溶剂中石油醚和乙酸乙酯的体积比依次为100:1、80:1、50:1、20:1、10:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到a
–
g共7个组分。
[0070]
步骤(5)中,浸膏干法上样的具体方法为:将组分b用10倍体积甲醇溶解后,用组分b等质量80目硅胶吸附拌样后上样;
[0071]
所述的正相硅胶柱中装柱硅胶为100目的正相硅胶;
[0072]
梯度洗脱采用的流动相为石油醚和乙酸乙酯,石油醚和乙酸乙酯的体积比依次为100:1、60:1、30:1、10:1、1:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到b1
–
b5共5个亚组分。
[0073]
步骤(6)中,所述的纯化先采用经凝胶色谱纯化,再采用hplc纯化。
[0074]
步骤(6)中,凝胶色谱纯化采用的凝胶柱为sephadex lh-20,以体积比为1:1的二氯甲烷和甲醇的混合溶剂作为流动相,然后进行hplc;
[0075]
所述的hplc色谱条件为流动相为体积比为19:1甲醇和水的混合溶剂,流速为1ml/min,收集开始出峰时间为5.6min的组分,采用安捷伦c
18
反向色谱柱,规格为:4.6mmx250nm,5um;柱温:30℃,每次进样量:20ul,采用紫外检测器,检测波长为210nm。
[0076]
实施例4
[0077]
二倍半萜类化合物的制备方法包括如下步骤:
[0078]
步骤(1),将真菌emericella sp.xl029使用大米固体培养基进行培养,静置培养30天,得到发酵产物;
[0079]
步骤(2),用甲醇萃取步骤(1)得到的发酵产物,将所得到的萃取液减压浓缩得到粗浸膏;
[0080]
步骤(3),将步骤(2)得到的粗浸膏用水溶解后,用乙酸乙酯反萃,取乙酸乙酯相减压浓缩,得到浸膏;
[0081]
步骤(4),将步骤(3)得到的浸膏干法上样至正相硅胶柱,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到 7个组分a
–
g;
[0082]
步骤(5),将步骤(4)得到的组分b干法上样至正相硅胶柱层析,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到5个亚组分b1
–
b5;
[0083]
步骤(6),对步骤(5)得到的亚组分b1进行纯化,得到式(ⅰ)所示的二倍半萜类化合物。
[0084]
步骤(2)中,所述的萃取方式是加入等体积甲醇在室温条件下超声1.1h,然后室温条件下静置11h,取上清,减压浓缩;萃取次数为4次。
[0085]
步骤(3)中,所用的水和乙酸乙酯的体积相同。
[0086]
步骤(4)中,浸膏干法上样的具体方法为:将浸膏用10倍体积甲醇溶解后,用浸膏等质量100目硅胶吸附拌样后上样;
[0087]
所述的正相硅胶柱中装柱硅胶为200目的正相硅胶;
[0088]
梯度洗脱采用的流动相按洗脱顺序依次为石油醚、石油醚和乙酸乙酯的混合溶剂、甲醇,石油醚和乙酸乙酯的混合溶剂中石油醚和乙酸乙酯的体积比依次为100:1、80:1、50:1、20:1、10:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到a
–
g共7个组分。
[0089]
步骤(5)中,浸膏干法上样的具体方法为:将组分b用10倍体积甲醇溶解后,用组分b等质量100目硅胶吸附拌样后上样;
[0090]
所述的正相硅胶柱中装柱硅胶为200目的正相硅胶;
[0091]
梯度洗脱采用的流动相为石油醚和乙酸乙酯,石油醚和乙酸乙酯的体积比依次为100:1、60:1、30:1、10:1、1:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到b1
–
b5共5个亚组分。
[0092]
步骤(6)中,所述的纯化先采用经凝胶色谱纯化,再采用hplc纯化。
[0093]
步骤(6)中,凝胶色谱纯化采用的凝胶柱为sephadex lh-20,以体积比为1:1的二氯甲烷和甲醇的混合溶剂作为流动相,然后进行hplc;
[0094]
所述的hplc色谱条件为流动相为体积比为19:1甲醇和水的混合溶剂,流速为1ml/min,收集开始出峰时间为5.6min的组分,采用安捷伦c
18
反向色谱柱,规格为:4.6mmx250nm,5um;柱温:30℃,每次进样量:20ul,采用紫外检测器,检测波长为210nm。
[0095]
实施例5
[0096]
二倍半萜类化合物的制备方法包括如下步骤:
[0097]
步骤(1),将真菌emericella sp.xl029使用大米固体培养基进行培养,静置培养29天,得到发酵产物;
[0098]
步骤(2),用甲醇萃取步骤(1)得到的发酵产物,将所得到的萃取液减压浓缩得到粗浸膏;
[0099]
步骤(3),将步骤(2)得到的粗浸膏用水溶解后,用乙酸乙酯反萃,取乙酸乙酯相减压浓缩,得到浸膏;
[0100]
步骤(4),将步骤(3)得到的浸膏干法上样至正相硅胶柱,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到 7个组分a
–
g;
[0101]
步骤(5),将步骤(4)得到的组分b干法上样至正相硅胶柱层析,然后进行梯度洗脱,收集各梯度的梯度洗脱液并浓缩,经tlc监测,合并相同的部分,得到5个亚组分b1
–
b5;
[0102]
步骤(6),对步骤(5)得到的亚组分b1进行纯化,得到式(ⅰ)所示的二倍半萜类化合物。
[0103]
步骤(2)中,所述的萃取方式是加入等体积甲醇在室温条件下超声1h,然后室温条件下静置10h,取上清,减压浓缩;萃取次数为3次。
[0104]
步骤(3)中,所用的水和乙酸乙酯的体积相同。
[0105]
步骤(4)中,浸膏干法上样的具体方法为:将浸膏用10倍体积甲醇溶解后,用浸膏等质量90目硅胶吸附拌样后上样;
[0106]
所述的正相硅胶柱中装柱硅胶为160目的正相硅胶;
[0107]
梯度洗脱采用的流动相按洗脱顺序依次为石油醚、石油醚和乙酸乙酯的混合溶剂、甲醇,石油醚和乙酸乙酯的混合溶剂中石油醚和乙酸乙酯的体积比依次为100:1、80:1、50:1、20:1、10:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到a
–
g共7个组分。
[0108]
步骤(5)中,浸膏干法上样的具体方法为:将组分b用10倍体积甲醇溶解后,用组分b等质量90目硅胶吸附拌样后上样;
[0109]
所述的正相硅胶柱中装柱硅胶为160目的正相硅胶;
[0110]
梯度洗脱采用的流动相为石油醚和乙酸乙酯,石油醚和乙酸乙酯的体积比依次为100:1、60:1、30:1、10:1、1:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到b1
–
b5共5个亚组分。
[0111]
步骤(6)中,所述的纯化先采用经凝胶色谱纯化,再采用hplc纯化。
[0112]
步骤(6)中,凝胶色谱纯化采用的凝胶柱为sephadex lh-20,以体积比为1:1的二氯甲烷和甲醇的混合溶剂作为流动相,然后进行hplc;
[0113]
所述的hplc色谱条件为流动相为体积比为19:1甲醇和水的混合溶剂,流速为1ml/min,收集开始出峰时间为5.6min的组分,采用安捷伦c
18
反向色谱柱,规格为:4.6mmx250nm,5um;柱温:30℃,每次进样量:20ul,采用紫外检测器,检测波长为210nm。
[0114]
应用实例1
[0115]
该二倍半萜类化合物quiannulatic acid的制备方法,包括如下步骤:
[0116]
1)将真菌emericella sp.xl029使用大米固体培养基进行培养,静置培养30天,发酵产物为本发明使用的材料;
[0117]
2)用甲醇萃取上述发酵产物,减压浓缩得到粗浸膏约0.4kg;所述的萃取方式是加入等体积甲醇在室温条件下超声1h,然后室温条件下静置3h,取上清,减压浓缩;萃取次数为3次。
[0118]
3)将上述粗浸膏用水溶解后用乙酸乙酯反萃,水和乙酸乙酯体积比为1:1,取乙酸乙酯相减压浓缩,得到浸膏约0.1kg;
[0119]
4)上述浸膏使用甲醇溶解后用硅胶(ca.0.2kg)吸附拌样,经正相硅胶柱色谱粗分梯度洗脱得到组分a
–
g,硅胶柱层析的固定相为100-200目的正相硅胶,流动相依次为石油醚,二氯甲烷/甲醇(100:0
→
0:100);
[0120]
具体为:将浸膏用10倍体积甲醇溶解后,用浸膏等质量100目硅胶吸附拌样后上样;
[0121]
梯度洗脱采用的流动相按洗脱顺序依次为石油醚、二氯甲烷和甲醇的混合溶剂、甲醇,二氯甲烷和甲醇的混合溶剂中二氯甲烷和甲醇的体积比依次为10 0:1、80:1、50:1、20:1、10:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到a
–
g共7个组分。
[0122]
5)组分b经正相硅胶柱层析,被分成5个亚组分(b1
–
b5),硅胶柱层析的固定相为
100-200目的正相硅胶,流动相为石油醚/乙酸乙酯(100:1
→
1:1);
[0123]
具体为:将组分b用10倍体积甲醇溶解后,用组分b等质量100目硅胶吸附拌样后上样;梯度洗脱采用的流动相为石油醚和乙酸乙酯,石油醚和乙酸乙酯的体积比依次为100:1、60:1、30:1、10:1、1:1,每个梯度洗脱到tlc点板无点后,更换下一梯度洗脱,得到b1
–
b5共5个亚组分。
[0124]
6)b1经凝胶柱层析sephadex lh-20(meoh),hplc纯化,凝胶的流动相为二氯甲烷/甲醇(1:1),hplc的流动相为甲醇/水(19:1),流速为1ml/ min,收集开始出峰时间为5.6min的组分,得到化合物quiannulatic acid(1. 2mg)。
[0125]
凝胶色谱纯化时,将亚组分b1用2倍体积甲醇溶解后上样。
[0126]
使用10ml试管进行收集,每一管收集6ml,本次共收集60管,tlc(薄层色谱)分析后,经tlc监测,合并相同的部分,将10-20管进行合并,减压浓缩后经hplc纯化。
[0127]
所述的hplc色谱条件为流动相为体积比为19:1甲醇和水的混合溶剂,流速为1ml/min,收集开始出峰时间为5.6min的组分,采用安捷伦c
18
反向色谱柱,规格为:4.6mmx250nm,5um;柱温:30℃,每次进样量:20ul,采用紫外检测器,检测波长为210nm。
[0128]
通过核磁确定了获得的单体化合物的结构,所得化合物的性状和波谱数据如下:
[0129]
本发明二倍半萜类化合物结构式如式(ⅰ)所示;
[0130][0131]
中文名如何命名,英文名为quiannulatic acid,淡黄色无定型粉末,分子式为c
25h38
o2,分子量为:370.2872。
[0132]1h nmr(cdcl3,600mhz)δh:2.90(1h,d,j=15.80hz,h-18a); 2.40(1h,t,j=9.10hz,h-7);2.19(1h,d,j=15.80hz,h-18b); 2.09(1h,m,h-2a);2.04(1h,m,h-6a);1.93(1h,m,h-20);1.88(1h, m,h-5);1.84(1h,m,h-3a);1.72(1h,m,h-9a),1.66(2h,m,h-10 a);1.57(1h,m,h-14a);1.52(1h,m,h-3b),1.45(1h,m,h-6b);1.4 1(3h,s,h-25);1.30(6h,m,h-2b,h-9b,h-11,h-12,h-13a,h-14b,), 1.11(2h,m,h-10b,h-13b);0.94(3h,d,j=6.20hz,h-23);0.91(3 h,d,j=6.50hz,h-22);0.90(3h,s,h-24);0.81(3h,d,j=6.60h z,h-21)。
[0133]
13
c nmr(cdcl3,150mhz)δc:184.0(c-19),135.8(c-16),133.8(c
‑ꢀ
17),65.1(c-12)62.7(c-8),60.6(c-15),54.6(c-1),53.2(c-7),46.0(c-5), 45.7(c-4),43.1(c-11),39.0(c-2),36.8(c-18),35.7(c-10),34.8(c-14),31. 2(c-3),29.2(c-13),29.1(c-9),29.0(c-20),28.2(c-6),24.2(c-22),21.6(c
ꢀ‑
21),21.5(c-24),20.0(c-23),9.5(c-25).
[0134]
应用实例2
[0135]
将实施例所得的二倍半萜类化合物quiannulatic acid对2株临床耐药细菌进行mic测定,采用二倍稀释法,具体步骤包括如下:
[0136]
1)临床标准耐药菌mic的测定:分别将所得到的化合物用dmso配制成 10mm的贮备
液。
[0137]
配制500ml mh肉汤(具体操作为:取mh(mueller-hinton)肉汤粉末 10.5g(海博,青岛),加蒸馏水至500ml,调整ph为7.4
±
0.2,121℃高压 15min,备用。
[0138]
将atcc 43300、atcc 51299作为测试菌株;在pda平板复苏测试菌株,接种血琼脂平板(百博,济南)上,挑取单个菌落,获得纯菌。利用麦氏比浊仪(梅里埃,法国)配制菌悬液,用质量浓度为0.9%的生理盐水稀释纯菌至麦氏浊度为0.5mc(相当于1.5x10
8 cells/ml)的菌悬液,然后再用mh肉汤1: 10倍稀释后再进行1:10稀释,得到2倍终浓度接种菌液。
[0139]
将化合物贮备液10mm,用dmso(二甲基亚砜)稀释10倍,得到浓度为1mm抗菌药物溶液。根据该化合物相对分子量为370,设定8、4、2、1、 0.5、0.125μg/ml之间一系列浓度梯度,用mh肉汤进行稀释。取无菌96孔板,第一孔至第六孔分别加入100μl浓度为8、4、2、1、0.5、0.125μg/ml 的化合物,然后在加入100μl配制好的菌悬液(即2倍终浓度接种菌液),第七孔为阴性对照,加入100μl mh肉汤和100μl灭菌的蒸馏水,第八孔为阳性对照,分别加入100μl mh肉汤和100μl菌悬液(即2倍终浓度接种菌液)。每个孔设置3个重复,将处理好的96孔板放置到37℃的恒温培养箱中用锡箔纸包上避光培养,20-24h观察。如果某浓度的化合物具有活性,则在该浓度对应的孔是澄清的,反之浑浊。24h发现4-8μg/ml的化合物组完全抑制,阴性对照长满孔的底部,阳性对照无生长。化合物的工作浓度为2μg/ml,即化合物对耐药细菌的完全抑制浓度为2μg/ml。
[0140]
经测试,实施例1所得化合物quiannulatic acid对耐甲氧西林金黄色葡萄球菌和万古霉素耐药粪肠球菌这2株临床耐药细菌的最小抑菌浓度mic均为2 μg/ml。由此可见,所述的二倍半萜类化合物quiannulatic acid在抗临床耐药细菌方面具有非常大的应用潜能。
[0141]
以上显示和描述了本发明的基本原理、主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1