路易斯酸催化重氮化合物活化模式的筛选方法
1.本发明涉及一种路易斯酸催化重氮化合物活化模式的筛选方法,具体涉及一种基于密度泛函理论揭示路易斯酸催化重氮化合物活化模式的方法。
背景技术:
2.作为有机合成中的一类重要的活性底物,重氮化合物能够参与多种类型的转化反应进而合成有价值的加成产物。重氮化合物参与转化反应通常需要先生成高活性的碳卡宾前驱体,其生成需要脱除一分子氮气。然而,对于重氮化合物的研究大都集中在过渡金属催化转化生成金属卡宾中间体。目前,路易斯酸催化重氮化合物的转化反应吸引了越来越多的关注,其活化模式与过渡金属催化下重氮化合物的活化模式又相差甚远,尽管众多的实验结果对路易斯酸催化的重氮化合物的活化模式的研究起到了极大的帮助;由于现有实验条件的限制,过渡态和中间体的结构很难被观测或者捕捉,因此仅依靠实验手段并不能揭示其活化模式。
技术实现要素:
3.本发明的主要目的在于提供一种路易斯酸催化重氮化合物活化模式的筛选方法,从而克服现有技术中的不足。
4.为实现前述发明目的,本发明采用的技术方案包括:
5.一种路易斯酸催化重氮化合物活化模式的筛选方法,包括如下步骤:
6.(1)基于以b(c6f5)3为催化剂催化2-重氮-2-苯乙酸甲酯和苯乙炔及苯乙烯反应的实验条件信息,构建催化剂及反应底物的计算模型,并采用密度泛函理论方法对所述计算模型进行结构优化得到催化剂及反应底物的最稳定构型;
7.(2)根据重氮化合物的结构特点及路易斯酸催化剂的催化特性设计多种活化模式,然后在步骤(1)所获催化剂及反应底物的最稳定构型的基础上,对每种活化模式中涉及到的反应中间体和过渡态进行模型构建,并对所构建的反应中间体模型、过渡态模型进行结构优化和过渡态结构的搜寻;
8.(3)绘制各活化模式的反应势能剖面图,其中选择催化剂b(c6f5)3与2-重氮-2-苯乙酸甲酯(r1)的能量和为参考零点,势能面上所有的能量数值均以此为参考,进而获得各活化模式的反应能垒;
9.(4)将各活化模式的反应能垒进行比较,选取其中反应能垒最小的活化模式作为所述路易斯酸催化重氮化合物的活化模式。
10.在一些实施方式中,步骤(1)中采用m06-2x/6-31g(d,p)方法及smd溶剂化模型对所述计算模型进行结构优化。
11.在一些实施方式中,步骤(1)中是以二氯乙烷为溶剂并在298k、1atm条件下进行结构优化,得到催化剂及反应底物的最稳定构型。
12.在一些实施方式中,步骤(1)中在获得催化剂及反应底物的最稳定构型之后,再升
高基组在相同条件下进行能量校正(m06-2x/6-311+g(d,p)/smd
dce
),保证计算结果的准确度。
13.在一些实施方式中,步骤(2)中是采用qstn和ts相结合的方法进行所述过渡态结构的搜寻。
14.在一些实施方式中,步骤(3)中所述催化剂b(c6fs)3与2-重氮-2-苯乙酸甲酯(r1)的能量和对应的能量为gibbs自由能。
15.与现有技术相比,本发明基于密度泛函理论揭示了路易斯酸催化重氮化合物的活化模式,在预测路易斯酸催化重氮化合物反应的反应机理中可以提供详细的分子结构信息和原子水平的解释,对理解路易斯酸催化的重氮化合物的转化反应有重要的理论参考价值。
附图说明
16.图1示出了本发明一典型实施例中催化剂及反应底物的最稳定构型。
17.图2示出了本发明一典型实施例中活化模式a中的过渡态和中间体结构。
18.图3示出了本发明一典型实施例中活化模式b中的过渡态和中间体结构。
19.图4示出了本发明一典型实施例中活化模式c中的过渡态和中间体结构。
20.图5示出了本发明一典型实施例中活化模式d中的过渡态和中间体结构。
21.图6示出了本发明一典型实施例中路易斯酸催化重氮化合物活化模式的反应势能剖面图。
具体实施方式
22.下面将结合附图及实施例进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。
23.本实施例以b(c6f5)3为催化剂催化2-重氮-2-苯乙酸甲酯和苯乙炔及苯乙烯反应的反应为例,提供了一种基于密度泛函理论筛选路易斯酸催化重氮化合物活化模式的方法包括如下步骤:
24.(1)构建路易斯酸催化剂和重氮化合物的计算模型:
25.b(c6f5)3催化的2-重氮-2-苯乙酸甲酯和苯乙烯及苯乙炔的[2+1]环化反应参考下式:
[0026][0027]
基于以b(c6fs)3为催化剂催化2-重氮-2-苯乙酸甲酯和苯乙炔及苯乙烯反应的实验条件信息,构建催化剂、反应底物的计算模型;采用密度泛函理论方法对构建的模型进行结构优化得到催化剂、反应底物的最稳定构型。考虑到计算成本,结构优化采用的是m06-2x/6-31g(d,p)方法,溶剂模型选择的是smd溶剂化模型以二氯乙烷(dce)为溶剂在298k和
1atm下进行结构优化得到催化剂及反应底物的最稳定构型(如图1所示),然后再升高基组在相同条件下进行能量校正(m06-2x/6-311+g(d,p)/smd
dce
),保证计算结果的准确度。
[0028]
(2)反应路径设计:
[0029]
根据重氮化合物的结构特点及路易斯酸催化剂的催化特性,可以设计多种可能的活化模式,例如如下所示的modea、mode b、mode c、mode d四种活化模式,然后在步骤(1)优化得到的最稳定的催化剂和反应底物模型的基础上对每种可能的活化模式中涉及到的反应中间体和过渡态进行模型构建并对其进行结构优化和过渡态结构的搜寻,过渡态的搜寻使用的是qstn和ts相结合的方法。
[0030][0031]
a)构建活化模式a(mode a)中各反应物中间体、过渡态的模型:
[0032]
中间体建模:2-重氮-2-苯乙酸甲酯直接消除n2之后生成碳卡宾中间体a1,因此在建模的时候可在2-重氮-2-苯乙酸甲酯结构的基础上删除n2;碳卡宾中间体与路易斯酸b(c6f5)3结合有两种可能的模式,一种是与羰基氧配位另一种是与卡宾碳配位生成碳卡宾中间体a2和a3,建模时,分别连接o-b和c-b调整相应的距离分别为和然后设置计算参数[#opt freq m062x/6-31g(d,p)scrf=(smd,solvent=dichloroethane)]后进行结构优化得到相应的中间体的结构;
[0033]
过渡态建模:使用qst2的方法设置,即需要两个结构,其中一个为反应物另外一个为产物,产物中c-n之间的距离调整为左右,然后设置计算参数[#opt=qst2 freq m062x/6-31g(d,p)scrf=(smd,solvent=dichloroethane)];结构优化后得到优化后的过渡态结构,根据频率分析判断振动方向符合c-n断裂,证明所得过渡态结构为正确的结构。
[0034]
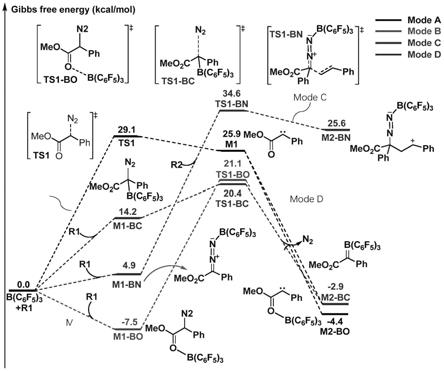
该活化模式a中的过渡态、中间体结构分别如图2中的ts1、m1所示。
[0035]
b)构建活化模式b(mode b)中各中间体、过渡态的模型:
[0036]
中间体建模:该活化模式中,路易斯酸b(c6f5)3首先与2-重氮-2-苯乙酸甲酯的羰基氧结合生成络合物中间体b1,调整b-o之间的距离为然后在此基础上消除n2之后
生成路易斯酸活化的碳卡宾中间体a2,设置参数与a)中相同,然后进行结构优化得到该活化模式下中间体的稳定构型;
[0037]
过渡态建模:在优化得到的络合物中间体基础上调整c-n键的距离为并把此距离固定,然后进行结构优化[计算参数:#opt=redundant freq m062x/6-31g(d,p)scrf=(smd,solvent=dichloroethane)],待优化完成后解除固定,使用ts方法搜寻该模式的过渡态,设置参数[#opt=(ts,calcfc)freq m062x/6-31g(d,p)scrf=(smd,solvent=dichloroethane)]进行结构优化,根据计算结果判断该结构即为所搜寻过渡态。
[0038]
该活化模式b中的过渡态、中间体结构分别如图3中的ts1-bo、m1-bo、m2-bo所示。
[0039]
c)构建活化模式c(mode c)中各中间体、过渡态的模型:
[0040]
中间体建模:该活化模式中,路易斯酸b(c6f5)3则与2-重氮-2-苯乙酸甲酯的氮原子结合生成络合物中间体c1,调整b-n之间的距离为要注意的是,在该模式下不能消除n2生成卡宾中间体,因此考虑到直接与烯烃或炔烃反应,本实施例中以苯乙烯为例,构建中间体模型c2,设置参与a)中相同,然后进行结构优化得到该活化模式下中间体的稳定构型;
[0041]
过渡态建模:在优化得到的中间体c2基础上调整c-c键的距离为并把此距离固定,然后进行结构优化[计算参数:#opt=redundant freq m062x/6-31g(d,p)scrf=(smd,solvent=dichloroethane)],待优化完成后解除固定,使用ts方法搜寻该模式的过渡态,设置参数[#opt=(ts,calcfc)freq m062x/6-31g(d,p)scrf=(smd,solvent=dichloroethane)]进行结构优化,根据计算结果判断该结构即为所搜寻过渡态。
[0042]
该活化模式c中的过渡态、中间体结构分别如图4中的ts1-bn、m1-bn、m2-bn所示。
[0043]
d)构建活化模式d(mode d)中各中间体、过渡态的模型:
[0044]
中间体建模:该活化模式中,路易斯酸b(c6f5)3则与2-重氮-2-苯乙酸甲酯的碳原子结合生成络合物中间体d1,调整b-c之间的距离为设置参与a)中相同,然后进行结构优化得到该活化模式下中间体的稳定构型;
[0045]
过渡态建模:在优化得到的中间体d基础上调整c-n键的距离为并把此距离固定,然后进行结构优化[计算参数:#opt=redundant freq m062x/6-31g(d,p)scrf=(smd,solvent=dichloroethane)],待优化完成后解除固定,使用ts方法搜寻该模式的过渡态,设置参数[#opt=(ts,calcfc)freq m062x/6-31g(d,p)scrf=(smd,solvent=dichloroethane)]进行结构优化,根据计算结果判断该结构即为所搜寻过渡态。
[0046]
该活化模式d中的过渡态、中间体结构分别如图5中的ts1-bc、m1-bc、m2-bc所示。
[0047]
采用密度泛函方法对设计的各条反应路径上反应物、中间体、过渡态和产物,基于化学键的生成和断裂形式以及催化剂吸附情况进行几何结构调整,然后在m06-2x/6-31g(d,p)/smd
dce
计算水平下进行结构优化并得到各基元反应的gibbs自由能。对势能面上中间体和过渡态使用进行结构优化并计算频率得到稳定结构后,同时保证每个中间体没有虚频,过渡态有且只有一个虚频,并读取thermal correction to gibbs free energy=后的吉布斯热校正量;为得到更准确的能量,进一步在更高的计算水平m06-2x/6-311+g(d,p)/smd
dce
进行但点能校正;单点能量计算输入文件参数设置为:[#m062x/6-311+g(d,p)scrf=(smd,solvent=dichloroethane)],最终得到gaussian的单点计算的输出文件*.log,读取
log文件中total energy;将单点能量加上吉布斯热校正量得到每个结构的吉布斯自由能量;最后选择催化剂的能量为参考零点绘出相应的势能面图(如图6所示)。
[0048]
3)分析各活化模式的能垒
[0049]
在绘制各活化模式反应势能剖面图时,选择催化剂b(c6f5)3与2-重氮-2-苯乙酸甲酯(r1)的能量和为参考零点,势能面上所有的能量数值均以此为参考,讨论时所用的能量均为gibbs自由能,具体分析如下:
[0050]
活化模式a中,重氮化合物首先消除一分子n2生成碳卡宾中间体m1,该过程的反应能垒是29.1kcal/mol,形成的卡宾中间体m1的相对能量为25.9kcal/mol,随后中间体m1与催化剂络合形成卡宾中间体m2-bc和m2-bo,其相对能量分别为-2.9kcal/mol和-4.4kcal/mol。
[0051]
活化模式b中,催化剂b(c6f5)3首先与2-重氮-2-苯乙酸甲酯(r1)络合生成前驱体m1-bo,该过程为放热过程,生成的m1-bo的相对能量为-7.5kcal/mol;然后中间体m1-bo消除一分子n2,经过过渡态ts1-bo生成路易斯酸活化的卡宾中间体m2-bo。此活化模式的反应能垒为27.8kcal/mol。
[0052]
活化模式c中,催化剂吸附在n原子上形成络合物中间体m1-bn,该中间体的相对能量为7.9kcal/mol,表明该过程是一个吸热过程。由于改活化模式不能发生n2消除反应,因此考虑了直接与亲核试剂反应(以与苯乙烯的反应为例)即,中间体m1-bn与苯乙烯反应经过过渡态ts1-bn生成中间体m2-bn。该模式的反应能垒为34.6kcal/mol,同时生成中间体m1-bn的相对能量为25.6kcal/mol。
[0053]
活化模式d中,催化剂与2-重氮-2-苯乙酸甲酯(r1)碳原子结合首先生成中间体m1-bc,与模式c相似,该吸附过程也是一个吸热过程,中间体m1-bc的相对能量为14.2kcal/mol,随后经过过渡态ts1-bc生成卡宾中间体m2-bc;计算结果表明该过程的反应能垒为21.1kcal/mol。
[0054]
综上所述,四种活化模式的反应能垒分别为29.1kcal/mol、27.8kcal/mol、34.6kcal/mol、21.1kcal/mol,通过比较可以发现,模式d的反应能垒最低,仅为21.1kcal/mol;因此,活化模式d为最可能的活化模式,即催化剂首先与2-重氮-2-苯乙酸甲酯(r1)的碳原子,然后消除n2生成高活性的b-c卡宾中间体,进而与苯乙烯和苯乙炔发生相应的转化反应。
[0055]
本实施例采用密度泛函理论(dft)对以b(c6f5)3为催化剂催化2-重氮-2-苯乙酸甲酯活化模式进行研究,其中包含四种可能的活化模式(modea~mode d),路径d是最有利的活化模式(δg≠=20.4kcal/mo1)。计算结果表明,催化剂吸附到重氮化合物的碳中心生成b-c络合物中间体m1-bc,然后消除一分子n2等到硼卡宾中间体的活化模式是能量最占优势的反应路径,同时也是整个反应的速率决定步骤。本发明对路易斯酸b(c6f5)3催化体系进行量子化学计算,从原子水平上研究了催化体系催化重氮化合物的活化模式及其与催化剂的构效关系。通过对该催化体系反应机理进行探索,不仅可以观察反应过程,而且能够为设计新型高效催化剂提供理论基础。
[0056]
本发明通过量子化学技术手段从原子水平上获得反应机理和催化剂构效关系,并在此基础上建立了一种基于能量跨度模型计算b(c6f5)3为催化剂催化2-重氮-2-苯乙酸甲酯活化模式的方法,进而可以深入探究催化剂的作用,为设计高效新型催化剂提供有价值
的理论指导,帮助缩短催化剂的研发周期和降低经费投入。
[0057]
本发明的方法仅需要计算机即可完成,因此无需购买价格昂贵的大型设备或者高额的测试分析费用,不仅有利于降低催化剂的筛选成本更有利于反应底物扩展的速度。同时,计算结果与实验结果吻合,计算结果准确可靠。
[0058]
以上是结合实施例对本发明作详细说明,但是本发明的实施方式并不受上述实施例的限制,其它任何在本发明专利核心指导思想下所作的改变、替换、组合简化等都包含在本发明专利的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1