一种W/O/W型温敏栓塞剂的制备方法与流程
一种w/o/w型温敏栓塞剂的制备方法
技术领域
1.本发明涉及栓塞技术领域,更具体地说,是涉及一种w/o/w型温敏栓塞剂的制备方法。
背景技术:
2.经导管动脉栓塞化疗(trans-arterial chemo-embolization,tace)是目前中晚期肝癌的最重要疗法之一,栓塞剂在其中发挥着举足轻重的作用。以碘油乳液为代表的液体栓塞剂是目前应用最为广泛的肝肿瘤血管栓塞材料,基于碘油乳液的传统化疗栓塞被称为c-tace(convention-tace)。碘油乳液具有良好的流动性,可以顺利通过微导管,并实现肿瘤末梢血管栓塞,但是,碘油乳液的栓塞性较差,容易被血液冲刷代谢,发生复通,在这种情况下,往往需要搭配明胶海绵(gelation sponge)、pva等固体栓塞剂进行二次栓塞治疗,而进行二次栓塞治疗的操作繁琐,费用增加,有时还会引起栓塞并发症。
3.聚n-异丙基丙烯酰胺类温敏纳米凝胶在溶胶状态下具有低粘度,在人体温度环境下能够从良好的流动状态转变为不可流动的凝胶状态,兼具良好的流动性与栓塞性,能够克服传统栓塞剂的流动性与栓塞性之间的矛盾,同时还兼具载药缓释性能,有望成为新一代介入治疗用栓塞剂。该类温敏凝胶自身通常不具备显影功能,为实现术中可视,需要和显影剂共混。在实际手术过程中,往往通过选择增加显影剂用量来提高显影能力;但是,显影剂用量过大会导致凝胶粘度增大,流动性下降,同时还会导致凝胶强度下降而变得松软,影响栓塞效果。
技术实现要素:
4.有鉴于此,本发明的目的在于提供一种w/o/w型温敏栓塞剂的制备方法,以协调温敏栓塞剂的显影能力和栓塞效果。
5.本发明提供了一种w/o/w型温敏栓塞剂的制备方法,包括以下步骤:
6.提供温敏纳米凝胶,所述温敏纳米凝胶为具有三维网络结构的聚n-异丙基丙烯酰胺类聚合物,将所述温敏纳米凝胶和第一水性显影剂分散在水中,得到外水相;
7.提供油相,所述油相中分散有油性显影剂;
8.提供内水相,将所述内水相和所述油相混合,进行第一次乳化,得到w/o乳液;
9.将所述w/o乳液与所述外水相混合,在冰浴条件下进行第二次乳化,所述第二次乳化为间歇式乳化,得到w/o/w型温敏栓塞剂。
10.优选地,所述第二次乳化包括:在6000r/min~10000r/min转速下循环进行剪切乳化和停顿,循环次数至少为1,每次剪切乳化30s~60s,每次停顿30s~60s;
11.所述第二次乳化的总剪切时间为1min~10min。
12.优选地,所述第一次乳化包括:在6000r/min~10000r/min转速下剪切乳化1min~10min。
13.优选地,所述具有三维网络结构的n-异丙基丙烯酰胺类聚合物为n-异丙基丙烯酰
胺或n-异丙基丙烯酰胺与共聚单体的交联聚合物,所述共聚单体选自丙烯酸、n-正丙基丙烯酰胺、甲基丙烯酸、甲基丙烯酸羟乙酯、丙烯酸羟乙酯和丙烯酰胺中的一种或多种。
14.优选地,每100ml所述外水相中,所述温敏纳米凝胶的重量为1g~8g;和/或
15.所述温敏纳米凝胶和所述第一水性显影剂的质量比为(1~6):(0.5~50)。
16.优选地,所述油相的溶剂为碘油和/或注射用植物油。
17.优选地,所述油性显影剂和所述油相的溶剂为碘油,所述碘油和所述第一水性显影剂的质量比为(10~40):(0.5~50)。
18.优选地,所述内水相和所述油相的体积比为1:(1~8);和/或
19.以所述w/o/w型温敏栓塞剂的总质量为100份,所述温敏纳米凝胶的质量为1~6份。
20.优选地,将所述温敏纳米凝胶和第一水性显影剂分散在水中的步骤中,还加入有水性化疗药;所述水性化疗药与所述温敏纳米凝胶的质量比为(0.1~5):(1~6);和/或
21.将所述内水相和所述油相混合的步骤中,还加入有表面活性剂;所述表面活性剂与所述油相的质量比为(0.01~2):(10~40)。
22.优选地,所述油相中还分散有:油性化疗药;所述油性化疗药与所述油相的质量比为(0.05~1.5):(10~40);和/或
23.所述内水相还包括:水性化疗药物和/或第二水性显影剂。
24.本发明提供了一种w/o/w型温敏栓塞剂的制备方法,包括以下步骤:将温敏纳米凝胶和第一水性显影剂分散在水中,得到外水相;提供油相,油相中分散有油性显影剂;通过内水相,将内水相和油相混合,进行第一次乳化,得到w/o乳液;将w/o乳液与外水相混合,在冰浴条件下进行第二次乳化,第二次乳化为间歇式乳化,得到w/o/w型温敏栓塞剂。与现有技术相比,本发明一方面通过在冰浴条件下进行间歇式乳化,保证乳化过程在低临界转变温度(lcst)以下进行,避免剪切升温过高导致温敏纳米凝胶失去表面活性剂作用,以及避免温敏纳米凝胶发生凝胶相变而导致乳液破乳、分层,从而制得性能稳定的乳液;另一方面,通过采用特定步骤,实现整体较好的相互作用,得到的w/o/w型温敏栓塞剂外水相和油相中均可不同程度地添加有显影剂,通过调整外水相和油相中的显影剂用量可实现提高栓塞剂的显影能力的目的,同时,由于显影剂在外水相中的用量可调,使其在满足显影性能要求的同时,可有效避免因外水相中的显影剂用量过高而导致温敏纳米凝胶的流动性下降以及栓塞效果减弱的问题。此外,通过将油性显影剂分散在油相中,将水性显影剂分散在外水相中,使得不同极性的显影剂能够稳定存在于同一剂型中,有利于提高栓塞剂的稳定性。
25.另外,本发明提供的制备方法工艺简单,可调整性更强,能够充分溶解分散不同溶解性(水溶性、油溶性)的化疗药物,具有广谱载药性;同时本发明提供的制备方法得到的w/o/w型温敏栓塞剂由于具有温敏性,在常温下为液体,注射后转变为固体,能够有效解决现有栓塞剂所面临“流动性-栓塞性之间的矛盾”,具有广阔的应用前景。
附图说明
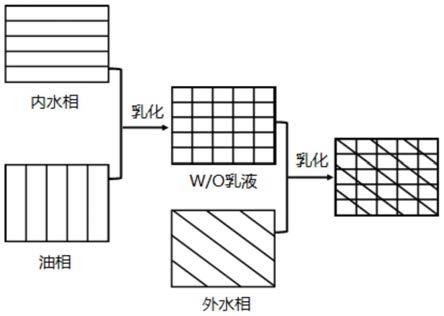
26.图1为本发明实施例提供的w/o/w型温敏栓塞剂的制备方法的工艺流程图;
27.图2为实施例1、对比例1~2提供的温敏栓塞剂进行x射线摄影的对比图。
具体实施方式
28.下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
29.本发明实施例提供了一种w/o/w型温敏栓塞剂的制备方法,包括以下步骤:
30.a)提供温敏纳米凝胶,温敏纳米凝胶为具有三维网络结构的聚n-异丙基丙烯酰胺类聚合物,将温敏纳米凝胶和第一水性显影剂分散在水中,得到外水相;
31.b)提供油相,油相中分散有油性显影剂;
32.c)提供内水相,将内水相和油相混合,进行第一次乳化,得到w/o乳液;
33.d)将w/o乳液与外水相混合,在冰浴条件下进行第二次乳化,第二次乳化为间歇式乳化,得到w/o/w型温敏栓塞剂。
34.可以理解的是,上述a)、b)、c)、d)并不限定本发明实施例各方法步骤的次序,可根据实际生产条件进行灵活调整步骤的次序,例如,步骤a)可在步骤b)和步骤c)完成后实施。
35.请参阅图1所示,图1为本发明实施例提供的w/o/w型温敏栓塞剂的制备方法的工艺流程图。
36.本发明首先将温敏纳米凝胶和第一水性显影剂分散在水中,得到外水相。
37.在本发明实施例中,温敏纳米凝胶为具有三维网络结构的聚n-异丙基丙烯酰胺类聚合物,优选为n-异丙基丙烯酰胺或n-异丙基丙烯酰胺与共聚单体的交联聚合物,更优选为n-异丙基丙烯酰胺与共聚单体的交联聚合物;该聚合物与常规的线性聚n-异丙基丙烯酰胺类聚合物不同,具有三维网络结构,记为聚n-异丙基丙烯酰胺类温敏纳米凝胶。
38.在本发明实施例中,共聚单体优选选自丙烯酸(aa)、n-正丙基丙烯酰胺(nnp)、甲基丙烯酸(maa)、甲基丙烯酸羟乙酯(hema)、丙烯酸羟乙酯(hea)和丙烯酰胺(aam)中的一种或多种;对应的,n-异丙基丙烯酰胺与共聚单体的交联聚合物包括交联剂交联的聚(n-异丙基丙烯酰胺)、交联剂交联的聚(nip-co-aa)、交联剂交联的聚(nip-co-nnp)、交联剂交联的聚(nip-co-mma)、交联剂交联的聚(nip-co-hema)、交联剂交联的聚(nip-co-hea)和交联剂交联的聚(nip-co-aam);交联采用的交联剂优选选自n,n'-亚甲基双丙烯酰胺、n,n'-亚乙基双丙烯酰胺、1,3-亚丙基双丙烯酰胺、二丙烯酸乙二酯、二乙二醇二丙烯酸酯、三乙二醇二丙烯酸酯、三羟甲基丙烷三丙烯酸酯、季戊四醇三丙烯酸酯中的一种或多种,更优选为n,n
’‑
亚甲基双丙烯酰胺。一些实施例中,交联剂为n,n'-亚甲基双丙烯酰胺(下文表示为mba)。
39.本发明对温敏纳米凝胶、共聚单体和交联剂的来源没有特殊限制,可采用本领域技术人员熟知的市售商品,也可以采用本领域常规技术手段进行制备获得的产物。
40.以mba交联的聚(nip-co-aa)为例,其制备方法优选具体为:
41.将n-异丙基丙烯酰胺,十二烷基硫酸钠,mba加入到装有回流冷凝管和导气装置的三颈瓶中,用超纯水在磁力搅拌下溶解,再往上述反应体系中通入高纯氮20min~40min,将反应体系加热到65℃~75℃,加入引发剂过硫酸钾,在n2气氛中、65℃~75℃下反应0.5-1h,再加入丙烯酸继续反应4~5h,得到白色浑浊悬浮液,将此悬浮液在超纯水中透析纯化后冻干,冻干粉即为聚(nip-co-aa)。
42.在本发明实施例中,第一水性显影剂优选选自碘海醇、碘帕醇、碘克沙醇和碘氟醇中的一种或多种,更优选为碘海醇、碘帕醇、碘克沙醇或碘氟醇。本发明对水性显影剂的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
43.步骤a)中,将温敏纳米凝胶和第一水性显影剂分散在水中的操作可参考本领域技术人员的常规操作,使得温敏纳米凝胶和第一水性显影剂完全溶解在水中即可,例如,可采用机械搅拌的方法。
44.将温敏纳米凝胶和第一水性显影剂分散在水中的步骤中,调整温敏纳米凝胶的添加量,使得每100ml外水相中的温敏纳米凝胶的质量为1g~8g;另外,还调整第一水性显影剂的添加量,使得温敏纳米凝胶和第一水性显影剂的质量比为(1~6):(0.5~50),如此,保证凝胶相变行为正常行使,避免由于第一水性显影剂的添加量过大或过小带来的负面影响,当第一水性显影剂的添加量小于上述添加量的下限值时,无法有效提升栓塞剂的显影能力;当第一水性显影剂的添加量大于上述添加量的上限值时,栓塞剂的栓塞效果下降。
45.在本发明实施例中,以w/o/w型温敏栓塞剂的总质量为100份计,温敏纳米凝胶的质量为1~6份,更优选为2.8~5.9份;在此基础上,保证栓塞剂具有良好栓塞效果的同时,还具有良好的流动性。
46.在本发明实施例中,以w/o/w型温敏栓塞剂的总质量为100份计,第一水性显影剂的质量为0.5~50份,更优选为1.12~30份。
47.在本发明实施例中,优选地,将温敏纳米凝胶和第一水性显影剂分散在水中的步骤中,还加入有水性化疗药;水性化疗药与温敏纳米凝胶的质量比为(0.1~5):(1~6)。更优选地,水性化疗药与温敏纳米凝胶的质量比为(0.5~3):(1~6)。
48.本发明实施例对水性化疗药物的具体种类和来源没有特殊限制,主要选为购买得到的药品。
49.本发明采用上述特定含量组成的外水相,通过将特定水性显影剂分散在外水相中,一方面,可协同油相部分的油性显影剂,达到增加制剂显影能力的目的,另一方面,外水相中的水性显影剂用量可调,可避免因外水相中的水性显影剂用量过高而影响栓塞剂的流动性及其凝胶相变行为,保证温敏栓塞剂的栓塞效果。值得注意的是,通过调整温敏栓塞剂中的温敏纳米凝胶和显影剂的用量在上述范围内,可保证制剂具有优异的栓塞效果和显影能力。
50.步骤b)中,提供油相,油相中分散有油性显影剂。
51.在本发明实施例中,油相的溶剂为油溶性溶剂,将油性显影剂分散在油溶性溶剂中,相容性好,稳定性高。油相的制备方法可参考本领域的常规技术,例如将油性显影剂加入油溶性溶剂中,搅拌均匀,即得。
52.一些实施例中,油相的溶剂为碘油和/或注射用植物油。优选地,油性显影剂和油相的溶剂为碘油,碘油和第一水性显影剂的质量比为(10~40):(0.5~50)。碘油既作为油相基质又作为显影剂,可保证最大化栓塞剂中油性显影剂的用量;而且,通过形成w/o/w型,使得水溶性的聚n-异丙基丙烯酰胺类温敏纳米凝胶能够与碘油稳定共存,实现在提高栓塞效果的同时显著提升制剂的稳定性。此外,通过调整碘油的添加量,使得碘油和第一水性显影剂的质量比为(10~40):(0.5~50),更优选为(20~34):(0.5~50),保证制剂稳定性,防止碘油过量导致破乳。
53.一些实施例中,油相中还分散有油性化疗药物,油性化疗药与碘油的质量比为(0.05~1.5):(10~40)。通过在油相中添加油性化疗药物,使其与碘油共同作为油相;同时,调整油性化疗药物,使得油性化疗药与碘油的质量比为(0.05~1.5):(10~40),优选为(0.07~1):(10~40)。
54.本发明实施例对油性化疗药物的具体种类和来源没有特殊限制,主要为购买得到的药品。
55.步骤c)中,提供内水相,将内水相和油相混合,进行第一次乳化,得到w/o乳液。
56.本发明对混合的方式没有特殊限制,将内水相和油相混合的步骤采用本领域技术人员熟知的人工搅拌或机械搅拌的技术方案均可,目的是混合均匀。
57.一些实施例中,内水相包括:水性化疗药物和/或第二水性显影剂。本发明实施例对内水相中分散的第二水性显影剂的浓度没有特殊限制,采用本领域技术人员熟知的0到饱和浓度范围内的浓度值均可。本发明实施例在内水相中分散第二水性显影剂,有利于提高温敏栓塞剂中的显影剂用量,从而进一步提高x射线显影能力。
58.在本发明实施例中,第二水性显影剂优选选自碘海醇、碘帕醇、碘克沙醇和碘氟醇中的一种或多种,更优选为碘海醇、碘帕醇、碘克沙醇或碘氟醇。本发明对第二水性显影剂的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
59.一些实施例中,以w/o/w型温敏栓塞剂的总质量为100份计,第二水性显影剂的质量为0.1~5份,更优选为0.5~2份。
60.在本发明实施例中,水性化疗药物的具体种类和来源与上述技术方案中的相同,在此不再赘述。
61.在本发明实施例中,以w/o/w型温敏栓塞剂的总质量为100份计,内水相中的水性化疗药物的质量为0.05~3份。
62.在本发明实施例中,内水相和油相的体积比优选为1:(1~8),更优选为1:(2~3)。
63.在本发明实施例中,第一次乳化包括:在6000r/min~10000r/min转速下剪切乳化1min~10min。
64.在本发明实施例中,将内水相和油相混合的步骤中,还加入有表面活性剂;表面活性剂与碘油的质量比为(0.01~2):(10~40)。在此基础上,第一次乳化的过程优选具体为:将内水相、油相和表面活性剂混合后,在6000r/min~10000r/min转速下剪切乳化1min~10min,得到w/o乳液。
65.在本发明实施例中,表面活性剂优选为聚甘油缩合蓖麻醇酸酯或吐温。本发明对表面活性剂的来源没有特殊限制,采用本领域技术人员熟知的市售商品即可。
66.在本发明实施例中,以w/o/w型温敏栓塞剂的总质量100份为计,表面活性剂的质量为0.01~2份,更优选为0.67~1.14份。
67.步骤d)中,将w/o乳液与外水相混合,在冰浴条件下进行第二次乳化,第二次乳化为间歇式乳化,得到w/o/w型温敏栓塞剂。
68.本发明实施例在得到w/o乳液后,将得到的w/o乳液与外水相混合后,以进行第二次乳化。外水相与上述技术方案中的相同,在此不再赘述。同样的,本发明实施例对混合的方式没有特殊限制,采用本领域技术人员熟知的人工搅拌或机械搅拌的技术方案均可,目的是混合均匀。
69.在本发明实施例中,内水相和油相之和与外水相的体积比优选为1:(1~5),更优选为1:(1~2)。
70.本发明实施例将w/o乳液与外水相的混合物于冰浴条件下进行第二次乳化,第二次乳化为间歇式乳化。间歇式乳化是指乳化过程中存在停顿,停顿步骤结束之后继续乳化步骤。通过在冰浴条件下进行间歇式乳化,保证乳化过程在低临界转变温度(lcst)以下进行,避免剪切升温过高导致温敏纳米凝胶失去表面活性剂作用,以及避免温敏纳米凝胶发生凝胶相变而导致乳液破乳、分层,从而制得性能稳定的乳液。
71.一些实施例中,第二次乳化包括:在6000r/min~10000r/min转速下循环进行剪切乳化和停顿,循环次数至少为1,每次剪切乳化30s~60s,每次停顿30s~60s;第二次乳化的总剪切时间为2min~10min。通过采用该乳化条件进行乳化,形成w/o/w乳液,避免乳化剪切强度过高导致的乳液破乳、分层,同时避免乳化剪切强度过低导致的工艺时间过长,增加劳动成本。
72.一具体实施例中,将w/o乳液与外水相混合后,冰浴条件下,在6000r/min转速下剪切乳化30s,停顿30s,再剪切30s,停顿30s,然后再循环剪切乳化和停顿4次,使总剪切时间为3min,得到w/o/w型温敏栓塞剂。
73.在本发明实施例中,聚n-异丙基丙烯酰胺类温敏纳米凝胶在低临界转变温度(lcst)以下呈溶胶状态,当温度大于或等于lcst时变得疏水凝聚而呈固态,破坏乳液原有的亲水亲油平衡,进而导致乳液破乳、分层等不稳定的现象,同时也会导致温敏纳米凝胶丧失其表面活性剂作用。在此基础上,本发明实施例通过在冰浴条件下进行间歇式乳化,可保证乳化过程在低临界转变温度(lcst)以下进行,避免剪切升温过高导致温敏纳米凝胶失去表面活性剂作用,以及避免温敏纳米凝胶发生凝胶相变而导致乳液破乳、分层,从而制得性能稳定的乳液。
74.本发明提供的制备方法工艺简单,可调整性更强,能够充分溶解分散不同溶解性(水溶性、油溶性)的化疗药物,具有广谱载药性;同时本发明提供的制备方法得到的w/o/w型温敏栓塞剂由于具有温敏性,在常温下为液体,注射后转变为固体,能够有效解决现有栓塞剂所面临“流动性-栓塞性之间的矛盾”,具有广阔的应用前景。
75.可以理解的是,上述制备方法中提到的水性化疗药物以及油性化疗药物,其添加目的是为了提高化疗效果,当然也可以省略该水性化疗药物、油性化疗药物,不影响上述复合乳液的制备,上述药物包括但不限于阿霉素、盐酸阿霉素、紫杉醇、顺铂、卡铂、奥沙利铂、多西他赛、吉西他滨、丝裂霉素、长春新碱以及替尼类抗肿瘤药物等。
76.综上,本发明实施例提供的w/o/w型温敏栓塞剂的制备方法,包括以下步骤:将温敏纳米凝胶和第一水性显影剂分散在水中,得到外水相;提供油相,油相中分散有油性显影剂;通过内水相,将内水相和油相混合,进行第一次乳化,得到w/o乳液;将w/o乳液与外水相混合,在冰浴条件下进行第二次乳化,第二次乳化为间歇式乳化,得到w/o/w型温敏栓塞剂。与现有技术相比,本发明一方面通过在冰浴条件下进行间歇式乳化,保证乳化过程在低临界转变温度(lcst)以下进行,避免剪切升温过高导致温敏纳米凝胶失去表面活性剂作用,以及避免温敏纳米凝胶发生凝胶相变而导致乳液破乳、分层,从而制得性能稳定的乳液;另一方面,通过采用特定步骤,实现整体较好的相互作用,得到的w/o/w型温敏栓塞剂外水相和油相中均可不同程度地添加有显影剂,通过调整外水相和油相中的显影剂用量可实现提
高栓塞剂的显影能力的目的,同时,由于显影剂在外水相中的用量可调,使其在满足显影性能要求的同时,可有效避免因外水相中的显影剂用量过高而导致温敏纳米凝胶的流动性下降以及栓塞效果减弱的问题。此外,通过将油性显影剂分散在油相中,将水性显影剂分散在外水相中,使得不同极性的显影剂能够稳定存在于同一剂型中,有利于提高栓塞剂的稳定性。
77.另外,本发明提供的制备方法工艺简单,可调整性更强,能够充分溶解分散不同溶解性(水溶性、油溶性)的化疗药物,具有广谱载药性;同时本发明提供的制备方法得到的w/o/w型温敏栓塞剂由于具有温敏性,在常温下为液体,注射后转变为固体,能够有效解决现有栓塞剂所面临“流动性-栓塞性之间的矛盾”,具有广阔的应用前景。
78.为了进一步说明本发明,下面通过以下实施例进行详细说明。本发明以下实施例所用的原材料均为市售,其中聚甘油缩合蓖麻醇酸酯表示为pgpr。
79.实施例1
80.本发明实施例1按照表1的配方制备了一种温敏栓塞剂,温敏纳米凝胶为mba交联的pnip,其具体制备方法包括以下步骤:
81.(1)温敏纳米凝胶为n,n
’‑
亚甲基双丙烯酰胺交联的聚(n-异丙基丙烯酰胺),其制备方法如下:
82.将2.263g n-异丙基丙烯酰胺,0.032g十二烷基硫酸钠,0.032g mba加入到装有回流冷凝管和导气装置的250ml的三颈瓶中,用170ml超纯水在磁力搅拌下溶解,再往上述反应体系中通入高纯氮30min,将反应体系加热到70℃,加入引发剂过硫酸钾0.095g,在n2气氛中、70
±
1℃下反应4.5h,得到白色浑浊悬浮液,将此悬浮液在超纯水中透析纯化后冻干,收集冻干粉,即得。
83.(2)在水中溶解盐酸阿霉素,使其浓度为5mg/ml,将此溶液作为内水相;
84.在碘油中溶解紫杉醇,使其浓度为3mg/ml,将此溶液作为油相;
85.将上述内水相、油相按照表1中质量比混合,体积比为3:7,加入表面活性剂聚甘油缩合蓖麻醇酸酯(pgpr),使其占总体系比例2%(质量分数);充分搅拌混合后,8000r/min转速下剪切乳化5min,得到w/o乳液,作为分散相。
86.(3)按照表1中质量比配制外水相,作为连续相;
87.将上述分散相和连续相按照表1中质量比混合搅拌,其体积比为4:6;冰浴条件下,在6000r/min转速下剪切乳化30s,停顿30s,再剪切乳化30s,停顿30s,再循环剪切乳化和停顿4次,使总剪切时间为8min,即得到可显影可载药的温度敏感型栓塞剂。
88.表1
89.90.实施例2
91.本发明实施例2按照表2的配方制备了一种温敏栓塞剂,温敏纳米凝胶为n,n
’‑
亚甲基双丙烯酰胺交联的聚(nip-co-aa),其具体制备方法包括以下步骤:
92.(1)温敏纳米凝胶为n,n
’‑
亚甲基双丙烯酰胺交联的聚(nip-co-aa),其制备方法如下:
93.将2.263g n-异丙基丙烯酰胺,0.032g十二烷基硫酸钠,0.032g n,n
’‑
亚甲基双丙烯酰胺加入到装有回流冷凝管和导气装置的250ml的三颈瓶中,用170ml超纯水在磁力搅拌下溶解,再往上述反应体系中通入高纯氮30min,将反应体系加热到70℃,加入引发剂过硫酸钾0.095g,在n2气氛中、70
±
1℃下反应0.5h,再加入0.27g丙烯酸,继续反应4h,得到白色浑浊悬浮液,将此悬浮液在超纯水中透析纯化后冻干,收集冻干粉,即得。
94.(2)在水中溶解吉他西滨,使其浓度为5mg/ml,将此溶液作为内水相;
95.在碘油中溶解紫杉醇,使其浓度为3mg/ml,将此溶液作为油相;
96.将上述内水相、油相按照表2中质量比混合,体积比为3:7,加入表面活性剂聚甘油缩合蓖麻醇酸酯(pgpr),使其占总体系比例2%(质量分数);充分搅拌混合后,6000r/min转速下剪切乳化8min,得到w/o乳液,作为分散相。
97.(3)按照表2中质量比配制外水相,作为连续相;
98.将上述分散相和连续相按照表2中质量比混合搅拌,其体积比为4:6;冰浴条件下,在7000r/min转速下剪切乳化30s,停顿30s,再剪切乳化30s,停顿30s,再循环剪切乳化和停顿6次,使总剪切时间为4min,即得到可显影可载药的温度敏感型栓塞剂。
99.表2
[0100][0101]
实施例3
[0102]
本发明实施例3按照表3的配方制备了一种温敏栓塞剂,温敏纳米凝胶为n,n
’‑
亚甲基双丙烯酰胺交联的聚(nip-co-mma),其具体制备方法包括以下步骤:
[0103]
(1)按照表3配方称取温敏纳米凝胶。
[0104]
(2)按照表3配方,配制内水相和油相,将内水相和油相按比例进行混合,加入表面活性剂,充分搅拌混合后,9000r/min转速下剪切乳化9min,得到油包水乳液,作为分散相。
[0105]
(3)按照表3配方,配制外水相作为连续相;然后将步骤(2)制备的分散相和连续相按比例混合,在冰浴条件下,在9000r/min转速下剪切乳化30s,停顿30s,再剪切乳化30s,停顿30s,再循环剪切乳化和停顿2次,使总剪切时间为2min,即得。
[0106]
表3
[0107][0108]
实施例4
[0109]
本发明实施例4按照表4的配方制备了一种温敏栓塞剂,温敏纳米凝胶为n,n
’‑
亚甲基双丙烯酰胺交联的聚(nip-co-aam),其具体制备方法包括以下步骤:
[0110]
(1)按照表4配方称取温敏纳米凝胶。
[0111]
(2)按照表4配方,配制内水相和油相,将内水相和油相按比例进行混合,加入表面活性剂,充分搅拌混合后,7000r/min转速下剪切乳化10min,得到油包水乳液,作为分散相。
[0112]
(3)按照表4配方,配制外水相作为连续相;然后将步骤(2)制备的分散相和连续相按比例混合,在冰浴条件下,在8000r/min转速下剪切乳化30s,停顿30s,再剪切乳化30s,停顿30s,再循环剪切乳化和停顿12次,使总剪切时间为7min,即得。
[0113]
表4
[0114][0115][0116]
对比例1
[0117]
采用实施例1提供的制备方法制备得到温敏栓塞剂;区别在于:将碘油替换为注射用大豆油。
[0118]
对比例2
[0119]
采用实施例1提供的制备方法制备得到温敏栓塞剂;区别在于:外水相中不含水性显影剂,水性显影剂的用量补至水,使得外水相中的水占温敏栓塞剂总量的52.41%。
[0120]
测试例
[0121]
1、取实施例1、对比例1~2提供的温敏栓塞剂,进行x射线摄影,如图1所示,实施例1的x射线对比能力明显优于对比例1~2。
[0122]
2、取实施例1~4的温敏栓塞剂于37℃条件下贮存14天,发现各温敏栓塞剂的外水相部分在37℃下发生相变,由液体转变为固体,且在贮存14天后外观均一,不破乳,表明本发明提供的温敏栓塞剂具有良好的稳定性,可保证其注入人体后长时间保证稳定的栓塞效
果。
[0123]
3、取实施例1~4的温敏栓塞剂于-5℃条件下贮存7天,贮后发现各温敏栓塞剂的外观均一,不破乳,不过度增稠,流动性好,表明本发明提供的温敏栓塞剂的冷贮存稳定性合格。
[0124]
4、取实施例1~4提供的温敏栓塞剂,进行各项性能测试,测试结果如表5所示。其中,实施例2的温敏纳米凝胶的用量为2.80%,而水性显影剂的用量为8.63%,其水性显影剂的用量相对于实施例1来说较高,由此导致模量降低,栓塞效果下降,但是,其模量和散逸率仍在可接受的范围内。
[0125]
表5实施例1~4提供的温敏栓塞剂的各项性能数据
[0126][0127][0128]
所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1