一种基于机器学习的比移值预测方法
1.本发明涉及有机化学极性测定领域,具体涉及一种基于机器学习的比移值预测方法。
背景技术:
2.在有机化学领域中,化合物的极性常通过薄层色谱分析法进行实验测定,测定的相对极性大小由比移值(也称rf值)表示。然而,薄层色谱分析法对实验者的经验要求和操作水平都比较高,尤其是要挑选出合适的展开剂使获得的比移值不至于过大或者过小,这使得极性的测定成为一项耗时耗力的工作,需要反复试错。同时,世界各地的化学实验室中每天都产生海量的极性数据,这些数据都未能很好地利用起来,导致重复测定和资源浪费。
3.随着计算机技术的发展,机器学习算法被广泛地运用于大数据分析任务中。其中,xgboost、lightgbm、神经网络等方法在数据的回归与预测方面都有着亮眼的表现,具有快速、稳定的优点与强大的泛化能力。在化学领域,由于实验获取数据的成本相对较高,机器学习算法往往表现不佳。因此,如何克服上述问题,在有限的数据下,基于机器学习开发出一种比移值预测算法,从化合物的结构与性质快速准确地预测出其在给定展开剂下的比移值是一个亟待解决的问题。
技术实现要素:
4.本发明的目的在提供一种基于机器学习的比移值预测方法,以解决上述现有技术中存在的问题,利用有限的数据训练出比移值预测模型,能够快速准确地预测出有机化合物在给定展开剂下的比移值。为实现上述目的,本发明提供如下技术方案:
5.一种基于机器学习的比移值预测方法,包含如下步骤:
6.a.数据采集与清洗:采集化合物、展开剂和比移值数据并进行清洗,以获取完整、无重复、无异常值的极性数据集。
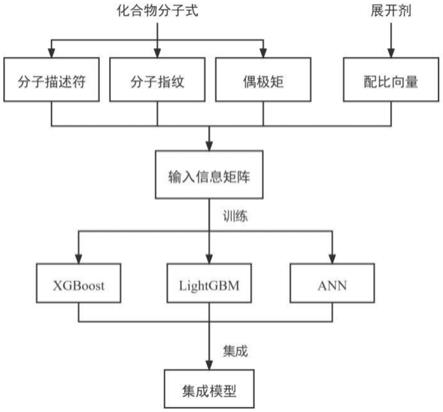
7.b.数据预处理:对清洗后的数据进行预处理,包括:将化合物的分子式数字化,通过分子指纹、偶极矩和分子描述符来表征化合物的分子结构与性质;使用展开剂配比向量表示展开剂体系及配比;预处理后的每条数据的信息向量包含多个维度,从而生成输入信息矩阵。
8.c.模型训练:将预处理后的数据划分为训练集和验证集,分别使用sigmoid函数约束的xgboost、lightgbm、神经网络算法在训练集上进行训练,将训练好的三个模型集成为一个集成模型用以预测比移值,在训练过程中利用验证集采用交叉验证的方法防止过拟合。
9.d.模型预测:利用步骤c训练好的比移值预测模型对目标化合物在目标展开剂体系下的比移值曲线进行预测,为实验测定提供指导。
10.e.反馈更新,根据实验反馈的数据自动更新和修正模型。
11.在上述步骤a中采集的数据包括:化合物的化学分子式,所使用的展开剂体系及配
比,以及化合物在该展开剂体系和配比下的比移值;清洗数据的方式包括:a)如果化合物的化学分子式,所使用的展开剂体系及配比,以及在该展开剂体系和配比下的比移值中有任意一个缺失,则去除该条数据;b)去除实验测定的比移值不合法的数据,即比移值大于1或者小于0的数据;c)去除重复数据,只保留其中一条或保留重复数据的均值。
12.上述步骤b中,所述分子指纹包含maccskeys指纹、morgan指纹等,视情况选择;所述分子描述符包含分子量、拓扑极性表面积、可旋转键的个数、氢键供体个数、氢键受体个数、脂水分配系数值等;所述偶极矩是指正、负电荷中心间的距离和电荷中心所带电量的乘积。如果涉及n种展开剂,则使用一个n维的向量表示展开剂体系及配比。
13.上述步骤c中,可以随机选取一些数据作为测试集,模型的预测能力由模型在测试集上的决定系数r2衡量,其计算公式如下:
[0014][0015]
其中n是测试集中样本的个数,yi是真实的比移值,是真实比移值的均值,是预测的比移值;r2越接近1代表模型的预测能力越强。
[0016]
步骤d对于要预测的目标化合物和目标展开剂体系及配比,先将其转化为输入信息矩阵,直接输入到训练好的集成模型中,即可得到预测的比移值。
[0017]
作为本发明的进一步方案,在利用训练好的比移值预测模型预测出比移值曲线后,模型将会根据曲线给出最优展开剂配比方案,使得实验获得的比移值不至于过大或者过小。
[0018]
作为本发明的进一步方案,对于多种不同的展开剂体系,展开剂的信息将以展开剂配比向量的形式输入至预测模型中。
[0019]
与现有技术相比,本发明的有益效果为:
[0020]
1、本发明通过机器学习方法建立比移值预测模型,能够快速准确地预测出目标化合物在目标展开剂体系下的比移值曲线,并根据曲线给出最优展开剂配比方案,使得实验获得的比移值不至于过大或者过小,极大地减少了薄层色谱分析技术对实验者经验的依赖,解决了展开剂选择的问题,避免了枯燥的重复实验,极大地提升了极性测定的效率,节省了时间和人力成本。
[0021]
2、本发明集成了sigmoid函数约束的xgboost、lightgbm、神经网络算法,利用比移值的值域在0和1之间的物理约束与多算法集成的技术,使得预测模型具有更强的稳定性和预测能力。
[0022]
3、本发明能够利用新的实验数据周期性更新修正原有的预测模型,将海量的极性数据有效利用起来,不断增强模型的泛化能力,避免了资源的浪费。
[0023]
4、本发明将机器学习技术引入实验化学领域,使得没有实验条件的情况下获取化合物的比移值成为一种可能,在实验化学、药物合成与分析等领域有着广泛应用和重要意义。
附图说明
[0024]
图1为本发明的比移值预测方法整体流程框图。
[0025]
图2为本发明的比移值预测方法中数据预处理与模型训练的流程框图。
[0026]
图3为本发明的一个实施例在预测集上的总体预测效果图。
[0027]
图4为本发明的具体实施效果图,即通过训练好的比移值预测模型预测出新化合物在目标展开剂体系下的比移值曲线。
具体实施方式
[0028]
下面将结合附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一个实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0029]
总体而言,本发明提出了一种基于机器学习的比移值预测方法,包含以下步骤:
[0030]
1)数据获取与清洗
[0031]
本实施例中,比移值数据的获取由自动化薄层色谱分析平台获得。进一步地,标准化的实验环境下产生的人工实验数据也可以作为有效数据获取。具体而言,获取的数据包括化合物的化学分子式,所使用的展开剂体系及配比以及在该体系和配比下的比移值。在本实施例中,共获取4928个数据,包含352个有机化合物在2种展开剂体系下的比移值,所述有机化合物的种类包括酮类、杂环类、醚类、卤化物类、醇类等。展开剂体系包括正己烷(hexane):乙酸乙酯(ea)体系与氯仿(dcm):甲醇(meoh)体系。对于hexane:ea体系,共有9种配比,包括1:0,50:1,20:1,5:1,3:1,1:1,1:2和0:1。对于dcm:meoh体系,共有5种配比,包括1:0,100:1,50:1,30:1,20:1和10:1。考虑到在实验过程中存在一些极端情况导致部分数据缺失,因此需要对数据进行清洗。具体清洗实施方式为:
[0032]
a)化合物的化学分子式,所使用的展开剂体系及配比以及在该体系和配比下的比移值中有任意一个缺失则去除该条数据。
[0033]
b)去除实验测定的比移值不合法的数据,即比移值大于1或者小于0的数据。
[0034]
c)去除重复数据,只保留其中一条或保留重复数据的均值。
[0035]
在数据清洗后,共有4114条数据合格。
[0036]
2)数据预处理
[0037]
在本实施例中,需要对清洗后的数据进行预处理,从而生成输入信息矩阵,如图2所示。由于化学分子式无法直接输入到计算机中,本实施例将其数字化为分子指纹、分子描述符和偶极矩来表征该化合物的结构与性质。具体而言,分子指纹包含maccskeys指纹、morgan指纹等,可视情况选择;分子描述符包含分子量、拓扑极性表面积、可旋转键的个数、氢键供体个数、氢键受体个数、脂水分配系数值等;偶极矩是正、负电荷中心间的距离和电荷中心所带电量的乘积,与物质的极性有着密切的联系,由计算化学手段得出。在本实施例中,使用了maccskeys分子指纹及偶极矩,分子量、拓扑极性表面积、可旋转键的个数、氢键供体个数、氢键受体个数、脂水分配系数值作为分子描述符。
[0038]
进一步地,由于存在多种常用的展开剂体系,本实施例使用展开剂配比向量表示展开剂的体系及配比。具体而言,展开剂的种类包括hexane、ea、dcm和meoh等,设此处考虑n
个展开剂,则使用一个n维的向量表示展开剂之间的比例关系。例如,[hexane,ea,dcm,meoh]=[0.75,0.25,0,0]表示展开剂的体系及配比为hexane:ea=3:1。在本实施例中,数据预处理后,每条数据的信息向量包含178个维度,其中分子指纹167个维度,展开剂配比向量4个维度,偶极矩和分子描述符共7个维度。整体而言,整个数据集为一个4114
×
178的信息矩阵。
[0039]
进一步地,数据预处理后将存储至专有的化合物极性数据库中,在周期性地接收新数据后,该数据库将自动更新。
[0040]
3)模型训练
[0041]
在本实施例中,采用基于sigmoid函数约束的xgboost、lightgbm和人工神经网络的集成算法作为训练模型。具体而言,sigmoid函数为:
[0042][0043]
考虑到存在比移值的值域在0和1之间这个物理约束,而sigmoid函数的值域也为(0,1),使用sigmoid函数作为物理约束可以提高模型预测的稳定性。xgboost、lightgbm和人工神经网络均可以通过python快速实现,并通过训练数据进行训练,设训练好的模型分别为g
xgb
,g
lgb
和g
ann
。则集成模型可表示为:
[0044][0045]
进一步地,在训练过程中,划分训练集和验证集,采用交叉验证的方法防止过拟合。在神经网络算法的训练过程中,使用提前终止,dropout等手段防止过拟合,提升模型的性能。在本实施例中,随机选取330个化合物在不同展开剂体系下的比移值作为训练集,共3372个数据,随机选取20个化合物在不同展开剂体系下的比移值作为验证集,共107个数据,随机选取10个化合物在不同展开剂体系下的比移值作为测试集,共112个数据。模型的预测能力由模型在测试集上的决定系数r2衡量,其计算公式如下:
[0046][0047]
其中n是测试集中样本的个数,yi是真实的比移值,是真实比移值的均值,是预测的比移值。r2越接近1代表模型的预测能力越强。在本实施例中,训练好的集成模型的r2为0.9469,这表明该模型具有很强的预测能力。在测试集上,该集成模型预测的比移值与真实比移值的散点图如图3所示,可以看出该模型的预测误差很小,散点基本处于y=x线上。
[0048]
优选地,本发明中的机器学习算法中的超参数的选取均可通过grid-search的方式寻找到最优参数组合。
[0049]
4)模型预测
[0050]
模型训练完毕后将使用训练好的模型进行预测。具体而言,对于要预测的目标化合物及目标展开剂体系与配比,先将其转化为输入信息矩阵,直接输入到训练好的集成模型中,即可得到预测的比移值。在本实施例中,两个新化合物分别在两种不同的展开体系下
的比移值曲线如图4所示。三角标号代表实验值,
×
标号代表预测值。由此可以表明,对于模型从未见过的新的化合物,本预测模型能够以高精度预测其在不同展开体系下的比移值曲线。
[0051]
进一步地,模型预测支持同时输入多条数据,将同时得出对应的比移值预测。
[0052]
进一步地,对于目标化合物,预测模型可以预测其在目标展开剂体系下的比移值曲线,根据曲线比移值在0.3~0.7之间所对应的展开剂配比,提供配置展开剂的指导。
[0053]
5)模型更新
[0054]
在本实施例中,预测模型将在原有模型的基础上周期性地加入新数据进行更新训练,以不断提高模型的泛化能力。具体而言,新数据的来源为实验反馈与自动化薄层色谱分析平台实验数据。进一步地,在每轮更新后将会使用测试集对模型的预测效果进行评估,评估合格则保留此次更新,不合格则回退至更新前的版本。具体而言,当模型预测的确定系数大于等于85%时,模型达标,反之则不达标。在本实施例中,每检测到数据库更新后就进行一次更新训练。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1