一种基于SNP易感位点的高度近视预测模型及其应用
一种基于snp易感位点的高度近视预测模型及其应用
技术领域
1.本发明属于生物医学领域,具体涉及一种基于snp易感位点的高度近视预测模型及其应用。
背景技术:
2.近视指在调节放松的状态下,平行光线经眼球屈光系统后聚焦在视网膜之前,在视网膜上不能清晰成像。屈光度为-6d(d指屈光度)或以上的近视眼为高度近视。高度近视是继白内障之后导致严重视力障碍和失明的第二大原因。高度近视患者患飞蚊症、斜视、视网膜脱离和眼底病变等并发症的风险明显高于普通近视。同时,高度近视的患者发生眼内血液循环障碍和组织变性的风险较高,更容易导致白内障、青光眼等并发症。近几十年来,高近视的发病率急剧增加,有研究推测,到2050年,高近视的人数可能达到9.38亿(占世界人口的9.8%)。研究结果显示,亚洲人群年轻人(6.8%~21.6%)高近视的发生率远高于非亚裔人群(2.0%~2.3%)。众所周知,遗传和环境可以促进近视的发展和进展,而高度近视更可能是遗传的。
3.近年来,随着基因组学和测序技术的发展,全基因组关联分析(gwas)已成为研究近视、高度近视的遗传易感性位点的主要方法。而单核苷酸多态性(snp)是一种广泛存在的基因组变异方式。snp是指某个人群中的正常个体中,在基因组dna的单个碱基对位置上存在不同的碱基的情况。在snp位点出现的碱基中,出现次数最少的称为最小等位,其频率称为最小等位频率(maf)。组成dna的碱基虽然有4种,但snp一般只有两种碱基组成,所以它是一种二态的标记,即二等位基因(biallelic)。由于snp的二态性,非此即彼,在基因组筛选中snps往往只需+/-的分析,而不用分析片段的长度,这就利于发展自动化技术筛选或检测snps。
4.已经有研究报道了与高度近视相关的snp位点,但大多未得到验证。而且,大多数已发表的研究都着眼于非亚洲人,他们的高近视发生率远低于亚洲人。由于遗传背景的差异,在非亚洲人群中鉴定的位点可能不会在亚洲人群中出现或具有不同的效果。
技术实现要素:
5.为开发适用于亚洲群体的高度近视诊断产品和方法,本发明对大量受试者进行了全外显子组测序(whole exome sequencing,wes)并对数据进行了全基因组病例对照关联研究,根据对参与者(9,852名高度近视患者和11,375名健康对照参与者)的测序数据的进行分析,本发明发现了新的易感snp位点并建立了高度近视的预测(诊断)模型,为临床预测(诊断)高度近视提供了新的依据。
6.第一方面,本发明提供了一组高度近视相关的snp位点组合,所述snp位点组合包含以下任意一种:
7.1)rs199970974、rs199873247、rs117708355、rs1228787360、rs41563615、rs9513522、rs9517546、rs533280354、rs16898116、rs866140198、rs76542212(本发明所述“模型1_1”、“模型1_2”、“模型1_3”的位点,共计11个);
8.2)rs199970974、rs199873247、rs117708355、rs1228787360、rs41563615、rs9513522、rs9517546、rs533280354、rs41558815、rs16898116、rs866140198、rs76542212、rs9264670、rs3094609、rs2001181、rs11554776、rs7380272、rs62626261、rs422951、rs2233580、rs520692、rs111265204、rs520803、rs72500812(本发明所述“模型2_1”或“模型2_2”的位点,共计24个);
9.3)rs199970974、rs199873247、rs117708355、rs1228787360、rs41563615、rs9513522、rs9517546、rs533280354、rs41558815、rs16898116、rs866140198、rs76542212、rs9264670、rs3094609、rs2001181、rs11554776、rs1065711、rs7380272、rs62626261、rs7380824、rs422951、rs2233580、rs520692、rs111265204、rs520803、rs72500812(本发明所述“模型2_3”的位点,共计26个);
10.4)以下第1-12个snp位点(本发明所述“模型1”的位点);
11.或以下第1-27个snp位点(本发明所述“模型2”的位点);
12.或以下第1-43个snp位点(本发明所述“模型3”的位点);
13.或以下第1-195个snp位点(本发明所述“模型4”的位点);
14.或以下第1-568个snp位点(本发明所述“模型5”的位点):
15.16.17.[0018][0019]
优选地,所述snp位点组合还包括以下snp位点中的至少一个或全部(297个)。
[0020]
[0021][0022]
本发明的所列出的snp位点从左至右,从上到下依次排列,具体是:第一行从左到右依次是第1-5个snp位点,第二行从左到右依次是第6-10个snp位点,依次类推。
[0023]
优选地,上述snp位点中“或”代表两个snp位点是该位点在不同版本dbsnp数据库中的名称,实质内容一致。例如第63个snp位点是rs1280768485或rs368234054,其中rs1280768485在染色体上的位置是190388283,在mrna上的位置是412,对应于第56个氨基酸的编码,该snp存在会导致所在基因表达的移码突变;其中rs368234054在染色体上的位置是190388285,在mrna上的位置是414,也对应于第56个氨基酸的编码,该snp同样会导致所在基因表达的移码突变,具体如下表1所示;因此rs1280768485或rs368234054可以互相替代。其他以“或”连接的snp位点于此同理。
[0024]
表1.rs1280768485和rs368234054信息对比
[0025]
dbsnp rs id染色体位置mrna位置氨基酸位置功能rs128076848519038828341256移码突变rs36823405419038828541456移码突变
[0026]
在本发明中,snp(单核苷酸多态性)是指dna中的单个碱基位置,受试者可以是纯合的或杂合的。本发明的snp位点以“rs
‑”
方式命名,本领域技术人员能够根据上文的rs-命
名,从适合的数据库和相关的信息系统如单核苷酸多态性数据库(dbsnp)中确定其确切的位置、核苷酸序列。
[0027]
另一方面,本发明提供了使用以上snp位点组合构建高度近视诊断模型的模型构建方法。
[0028]
优选地,所述方法使用的是逻辑回归的方法。
[0029]
优选地,本发明还包括10倍交叉验证的步骤。所述“10倍交叉验证”具体指利用sklearn.model_selection函数,选择0到100中的10个随机数字作为随机种子,分别将样本划分为10组训练集和验证集,进行10倍交叉验证,取10次结果的平均数作为模型结果。
[0030]
另一方面,本发明还提供了上述模型构建方法所构建的高度近视诊断模型;
[0031]
优选地,所述模型可以是公式、诺莫图、或其他方便受试者操作的方式;根据此模型,可以直接计算得到受试者当下是否是近视患者。
[0032]
另一方面,本发明提供了诊断高度近视的方法,所述方法包括根据本发明所述snp位点组合的检测结果判断高度近视的患病情况的步骤,或者,所述方法包括将snp位点组合的检测结果输入前述高度近视诊断模型得到判断结果的步骤。
[0033]
具体地,所述方法可以包括以下步骤:
[0034]
1)收集受试者样本,优选地,所述样本是口腔拭子(口腔试纸);
[0035]
2)对样本进行snp检测,优选地,所述检测还可以包括提取dna的步骤;
[0036]
3)根据2)的检测结果,判断受试者的高度近视患病情况;具体地,在受试者样本中检测到上述snp位点组合中至少1个、10个、100个、500个时代表该受试者是高度近视患者,或该受试者是高度近视的易感人群;所述snp位点的个数具体可以如本发明表4所示。
[0037]
所述判断可以是手动地、自动地、或它们组合地来执行或完成所选任务;可以根据检测结果手动计算结果,或输入前述系统自动地得到计算结果。
[0038]
另一方面,本发明提供了一种高度近视诊断的诊断系统,所述诊断系统包括使用上述snp位点组合或上述高度近视诊断模型进行计算的计算装置。
[0039]
优选地,所述诊断系统还可以包括snp位点的检测装置。
[0040]
另一方面,本发明提供了上述模型、系统、或检测上述snp位点组合的试剂在制备诊断近视的产品中的应用。
[0041]
优选地,所述检测上述snp位点组合的试剂包括但不限于以下方法检测snp时所使用的试剂:taqman探针法、测序法、芯片法、飞行质谱仪(maldi-tofms)检测、限制性片段长度多态性法(pcr-rflp)、单链构象多态性法(pcr-sscp)、等位基因特异性pcr(as-pcr)、snapshot法、snplex法、变性高效液相色谱法(dhplc)、变性梯度凝胶电泳法(dgge)。本领域技术人员可选择任一种或几种方法来检测snp位点,只要可以实现snp位点的检测。
[0042]
具体地,所述试剂包括但不限于引物、探针、芯片等。
附图说明
[0043]
图1是12个易感性snp的曼哈顿图。
[0044]
图2是12个snp的映射结果图。
[0045]
图3是go分析得到的热图。
[0046]
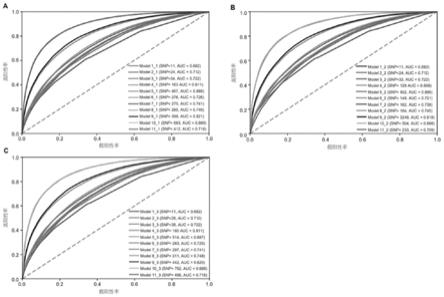
图4是本发明所提供的模型1~11在诊断高度近视中的roc曲线,a:模型1~5,b:模
型6~10,c:模型5、11、9。
[0047]
图5是经过特征筛选后的snp所构建的模型在诊断高度近视中的roc曲线,a:lasso筛选后构建的模型,b:linearsvc筛选后构建的模型,c:logistic regression筛选后构建的模型。
具体实施方式
[0048]
下面结合实施例对本发明做进一步的说明,以下所述,仅是对本发明的较佳实施例而已,并非对本发明做其他形式的限制,任何熟悉本专业的技术人员可能利用上述揭示的技术内容加以变更为同等变化的等效实施例。凡是未脱离本发明方案内容,依据本发明的技术实质对以下实施例所做的任何简单修改或等同变化,均落在本发明的保护范围内。
[0049]
实施例1、snp位点筛选
[0050]
本发明招募了21,227人(包括9,852名高度近视患者和11,375名健康对照)。根据标准程序从口腔粘膜样本(口腔试纸)中分离出所有受试者的基因组dna。
[0051]
本发明研究对象的排除标准是:
[0052]
有大于10%的snp缺失,平均测序深度低(《10),平均基因型质量低(《65),亲缘关系近(pihat》0.2),性别信息异常,非东亚血统,基因型个体的杂合度过高或者过低(杂合率平均值中偏离
±
4sd)
[0053]
snp位点的排除标准是:
[0054]
在个体中缺失率大于10%,明显偏离哈迪-温伯格平衡(p《1.0
×
10-6
),高度近视样本与对照样本的snp缺失率显著差异(》0.007),最小等位频率小于0.01
[0055]
经过以上筛选,来自20,955名个体(9,730名高近视患者和11,255名健康连续受试者)的89,268个snp(maf》0.01)用于随后的全基因组相关性分析。
[0056]
最后,p值《5.6
×
10-7
(=0.05/89,268)被认为具有统计学意义,相应的snp即为高度近视的易感性snp。共筛选到12个高度近视的易感snp,结果如图1所示。其中6个(50%)snp位点位于6号染色体上,表明6号染色体是高近视风险变异的主要富集区域。
[0057]
基因组膨胀因子λ是0.944,排除了由于群体分成造成的假阳性结果。将12个snp映射到距离最近的基因区域(表2、图2)。
[0058]
表2. 12个易感性snp详细信息
[0059]
[0060][0061]
这12个snp是高近视新发现的易感性snp,之前没有研究报道这些基因与高近视相关。在以往的报道中,这9个基因区域中:
[0062]
3个被报道过与屈光不正(refractive error)相关:dock9(rs9513522,rs9517546),znf204p(rs16898116),zscan9(rs76542212);4个被报道与眼科疾病相关:mdc1与白塞综合征(behcet syndrome)相关(pmid:20622878),7q11.2(rs1228787360)与角膜散光(corneal astigmatism)相关位点(pmid:29422769),hla-b(rs41563615,rs41558815)与史蒂文斯-约翰逊综合征(stevens-johnson syndrome)相关(omim:142830);hla-drb1被报道与葡萄膜脑膜炎综合征(uveomeningoencephalitic syndrome)(pmid:25108386)、干燥综合征(sjogren's syndrome)(pmid:28076899)相关。
[0063]
实施例2、连锁不平衡分析、snp的功能分析
[0064]
人类基因组中连锁不平衡(ld,linkage disequiibrium)的程度和分布在基因定位中起着重要作用,既可以作为复杂疾病精确定位的工具,也可以作为全基因组关联分析的基础。连锁不平衡映射(linkage disequiibrium mapping)的基本思想是检查整个基因组或候选基因附近的大量遗传标记位点,以找到似乎与疾病相关的位点,因为它们足够接近致病位点。本研究采用plink-block方法进行单体型块评估(haploid block estimation),鉴定具有高ld的易感snp。仅针对200kb以内的snp计算ld。
[0065]
通过连锁不平衡分析,发现了13个与以上12个snp高ld的snp位点;使用regulomedb v2.0和3dsnp v2.0对以上筛选的易感snp的功能进行研究。
[0066]
regulomedb数据库表明,12个snp中有8个排名得分在2b到4之间,有极强的证据证明它们可以影响转录调控。3dsnp v2.0数据库显示,在12个snp中,7个位于增强子组蛋白标记中,6个位于启动子组蛋白标记中,7个位于转录因子结合位点(tfbss),6个可以改变转录因子结合基序。这些结果表明,12个snp中的大多数可能对基因表达产生显著影响。
[0067]
gtex portal数据库显示5个snp(rs9513522,rs9517546,rs41563615,rs76542212,rs16898116)显着受影响的基因表达。rs9513522和rs9517546影响dock9和dock9-as2的表达。rs41563615影响多个基因的表达包括abcf1,c6orf15,cchcr1,hcg22,gtf2h4,hcg27,hla-b,hla-c,hla-s,ly6g5b,ly6g5c,mica,mir6891,pou5f1,psors1c1,psors1c2,psors1c3,tcf19,xxbac-bpg181b23.7,xxbac-bpg299f13.17.rs76542212影响al022393.9、linc01012、trim27、znf603p、zscan12、zscan23、z scan26和zscan31的表达。
rs16898116影响gusbp2的表达(表3)。
[0068]
表3. 12个易感snp的功能注释
[0069][0070]
实施例3、基因术语(go)和通路富集分析
[0071]
对12个易感性snp的注释基因进行go富集分析表明,共富集到82个显著的go术语(go terms)(p《0.05,补充表1),其中生物过程43个,细胞组分26个,分子功能13个。go富集是根据术语相似性对进行的聚类,使用的r包是simplifyenrichment。我们发现这些基因在免疫相关方面被富集,包括抗原加工呈递肽mhc,反应调节细胞免疫介导信号转导(图3)。为了证明基因与免疫之间的关联,我们通过输入"免疫"作为关键字,从amigo v2中收集了免疫go术语。然后,我们计算了基因的go术语和免疫go术语之间的相似性分数(similarity score),结果为0.685(p《0.0001)。
[0072]
共富集到32个kegg通路(p《《0.005,补充表1)涉及免疫相关通路,包括抗原加工和呈递,iga产生的肠道免疫网络和人类免疫缺陷病毒1的感染。
[0073]
实施例4、构建预测模型
[0074]
我们使用基于不同关联p值或不同来源下的snp的基因型进行逻辑预测建模。不同关联p值包括5.6
×
10-7
,1.0
×
10-5
,5.0
×
10-5
,1.0
×
10-3
,5.0
×
10-3
。基于以上标准,联合已经报道的snp位点,共获得了11组基于不同snp组合的模型;对每种snp组合进行不同方式的建模,每组snp组合都可以建立3中模型。
[0075]
1)获取snp基因图谱:利用python提取vcf中对应所需snps(共1354个snps),选择加性模型(未突变设置为0,突变一个等位为1,突变两个等位为2),获取原始的snp基因型谱;
[0076]
2)筛选snp基因图谱:计算原始snps基因型谱中每个样本中snps的缺失数目,如果缺失》5%(n_缺失/n_allsamples),去除对应的样本。对于缺失情况《5%的样本,利用人工神经网络进行补缺失。优选地,所述人工神经网络共三层,包括输入层,隐藏层以及输出层:输入层节点数为len(label_data),也就是完全没有缺失的snp;隐藏层设置20个节点;输出
层设置3个节点;设置学习率为0.003,训练迭代次数设置为20次;
[0077]
3)特征筛选:使用l1正则化的线性模型具有系数解,其许多估计系数为0。当目标是降低使用另一个分类器的数据集的维度,它们可以与feature_selection.selectfrommodel一起使用来选择非零系数。选择3种稀疏评估器包括用于回归的lasso以及用于分类的logistic regression和linearsvc
[0078]
4)构建模型:逻辑回归logisticregression方法构建模型,利用逻辑回归对样本进行分类,参数如下:
[0079][0080]
5)2次随机的10倍交叉验证:利用sklearn.model_selection函数,选择0到100中的10个随机数字作为随机种子,分别将样本划分为10组训练集和验证集,进行10倍交叉验证,取10次结果的平均数作为模型结果,以保证模型的客观性。
[0081]
省略步骤3)的情况下直接获得的模型命名为模型1-11,自身10倍交叉验证获取的平均auc结果图,如图4。
[0082]
对模型1~11进一步进行特征筛选,构建更精简的snp模型,更加方便检测和临床应用,具体以-1代表lasso筛选,-2代表linearsvc筛选,-3代表logistic regression筛选,也就是说模型1_1/2/3中的snp位点都选自模型1,以下同理。将筛选后的snp位点组合构建逻辑回归模型,计算auc值(如图5);汇总各模型的snp数量和auc数值如表4。
[0083]
表4.本发明所构建的模型及其效果验证
[0084]
[0085][0086]
注:表格中:lasso_l1表示用基于l1正则化的方法利用lasso筛选特征,然后构建逻辑回归模型;sv_l1表示基于l1正则化的方法利用linearsvc筛选特征,然后构建逻辑回归模型;log_l1表示基于l1正则化的方法利用logistic regression筛选特征,然后构建逻辑回归模型。
[0087]
本发明所述模型1具体指的是以下前12个snp位点。
[0088]
本发明所述模型2具体指的是以下前27个snp位点。
[0089]
本发明所述模型3具体指的是以下前43个snp位点。
[0090]
本发明所述模型4具体指的是以下前195个snp位点。
[0091]
本发明所述模型5具体指的是以下前568个snp位点。
[0092]
[0093]
[0094]
[0095][0096]
本发明所述模型6具体指的是模型1的snp位点联合以下297个snp位点共同构建的模型。
[0097]
本发明所述模型7具体指的是模型2的snp位点联合以下297个snp位点共同构建的模型。
[0098]
本发明所述模型8具体指的是模型3的snp位点联合以下297个snp位点共同构建的模型。
[0099]
本发明所述模型9具体指的是模型4的snp位点联合以下297个snp位点共同构建的模型。
[0100]
本发明所述模型10具体指的是模型5的snp位点联合以下297个snp位点共同构建的模型。
[0101]
以下列出snps_environment所代表的具体snp位点:
[0102]
[0103][0104]
本发明所述“模型1_1”、“模型1_2”、“模型1_3”具体指的是以下位点:rs199970974、rs199873247、rs117708355、rs1228787360、rs41563615、rs9513522、
rs9517546、rs533280354、rs16898116、rs866140198、rs76542212;
[0105]
本发明所述“模型2_1”或“模型2_2”具体指的是以下位点:rs199970974、rs199873247、rs117708355、rs1228787360、rs41563615、rs9513522、rs9517546、rs533280354、rs41558815、rs16898116、rs866140198、rs76542212、rs9264670、rs3094609、rs2001181、rs11554776、rs7380272、rs62626261、rs422951、rs2233580、rs520692、rs111265204、rs520803、rs72500812;
[0106]
本发明所述模型2_3具体指的是以下位点:rs199970974、rs199873247
[0107]
rs117708355、rs1228787360、rs41563615、rs9513522、rs9517546、rs533280354、rs41558815、rs16898116、rs866140198、rs76542212、rs9264670、rs3094609、rs2001181、rs11554776、rs1065711、rs7380272、rs62626261、rs7380824、rs422951、rs2233580、rs520692、rs111265204、rs520803、rs72500812。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1