颗粒和使用该颗粒的制剂的制作方法
1.本发明涉及以高含量包含原料药且粒径均匀的颗粒及使用该颗粒的制剂。
背景技术:
2.为了提高药物制剂的生产性,原料药(active pharmaceutical ingredient)与各种添加剂制粒。造粒法根据有无溶剂可分为湿式造粒和干式造粒。在对水不稳定的原料药进行造粒时,选择不使用溶剂的干式造粒法,在干式造粒法中,已知以热使添加剂熔化而用作粘合剂的熔融造粒法。
3.另一方面,熔融成分的物性对熔融造粒法的影响较大,因此很难控制造粒物的粒径。另外,由于熔融造粒法使用熔融成分来代替溶剂,因此很难提高造粒物中原料药的含有率,其结果必然会产生制剂大型化,并且服药依从性降低的问题。例如,专利文献1中记载了熔融造粒法制备的含药颗粒。但是,就专利文献1中记载的粒子而言,对于造粒粒子的控制没有进行任何研究。
4.(先行技术文献)
5.(专利文献)
6.专利文献1:日本特开第2016-511223号公报
技术实现要素:
7.(发明所要解决的问题)
8.本发明的目的之一是提供具有均匀粒径的熔融造粒物。此外,提供使用该熔融造粒物的药物组合物。
9.或者,本发明的目的之一是提供具有药物含量高的熔融造粒物。此外,提供使用该熔融造粒物的药物组合物。
10.(解决问题所采用的措施)
11.根据本发明的一个实施方式,提供了一种颗粒,该颗粒包括原料药、熔融成分和聚合物,其中,原料药、熔融成分和聚合物粘合(bind)在一起。
12.熔融成分在常温下是固体,可以具有100℃以下的熔点。
13.聚合物在常温下是固体,并且可以具有100℃以下的玻璃化转变点。
14.当熔融成分是硬脂酸或聚桂醇(lauromacrogol)时,聚合物可选自由甲基丙烯酸氨基烷基酯共聚物、甲基丙烯酸铵基烷基酯共聚物、甲基丙烯酸共聚物、羟丙甲纤维素乙酸酯丁二酸酯或聚乙烯吡咯烷酮组成的组。
15.可以具有原料药、熔融成分和聚合物熔融而相互混合的结构。
16.可以具有在原料药、熔融成分和聚合物中的一部分熔融而相互粘合的结构。
17.相对于原料药、熔融成分和聚合物的质量之和,可含有50质量%以上的原料药。
18.根据本发明的一个实施方式,提供包含上述任一种的颗粒和一种以上的药学上可允许的添加剂的制剂。
19.添加剂可以是崩解剂。
20.(发明的效果)
21.根据本发明的一个实施方式,提供具有高原料药含量和均匀粒径的颗粒。或者,根据本发明的一个实施方式,提供含有具有高原料药含量和均匀粒径的颗粒的制剂。
附图说明
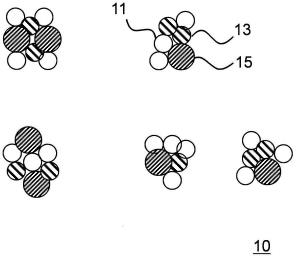
22.图1为示出本发明的一个实施方式的颗粒的示意图。
23.图2为说明本发明的一个实施方式的颗粒的制备方法的流程图。
24.图3为实施例1的颗粒的扫描型电子显微镜(sem)图像。
具体实施方式
25.以下,参照附图对本发明的颗粒和使用该颗粒的制剂进行说明。注意,本发明的颗粒和使用该颗粒的制剂不限于以下所示的实施方式和实施例所描述的内容。另外,在本实施方式和后述的实施例中参照的附图中,对相同部分或具有相同功能的部分赋予相同的标号,省略其重复的说明。
26.图1是示出本发明的实施方式之一的颗粒10的示意图(剖视图)。颗粒10包括原料药11、熔融成分13和聚合物15。颗粒10是将原料药11、熔融成分13和聚合物15通过熔融造粒来粘合而构成的粒子。在颗粒10中,原料药11没有特别限定。在制备颗粒10的方法中,由于不使用溶剂,特别是水,因此可以适当使用对水不稳定的原料药。
27.构成颗粒10的熔融成分13选自可适用于熔融造粒的油性添加剂。此外,熔融成分13优选选自不会因与原料药11的接触而使原料药11变性或类似物质显著增加的添加剂。为了通过熔融造粒法形成颗粒10,熔融成分13选自常温下为固体的添加剂。考虑到熔融造粒法通常使用的温度范围,熔融成分13优选选自熔点在100℃以下的添加剂,优选选自熔点在原料药11不会变性或类似物质不会明显增加的温度范围内的添加剂。此外,考虑与聚合物15的组合而选择熔融成分13。
28.聚合物15选自对于熔融成分具有相溶性并可适用于熔融造粒的添加剂。聚合物对熔融成分“具有相溶性”表示熔融成分与聚合物不分离的状态。或者,表示聚合物分散在熔融成分中或熔融成分分散在聚合物中的状态。在一个实施方式中,熔融成分和聚合物不分离的状态可以通过熔融成分和聚合物混合并熔化该熔融成分时的混合物(液体或具有流动性的半固体)的粘度的增加来确定。此外,聚合物15优选选自不会因与原料药11的接触而原料药11变性或类似物质显著增加的添加剂。为了通过熔融造粒法形成颗粒10,聚合物15选自常温下为固体的添加剂。考虑到熔融造粒法通常使用的温度范围,聚合物15优选选自玻璃化转变点(glass transition point)在100℃以下的添加剂,优选选自玻璃化转变点在原料药11不会变性或类似物质不会明显增加的温度范围内。此外,考虑与熔融成分13的组合而选择聚合物15。
29.作为这种熔融成分13和聚合物15的组合,当熔融成分13为硬脂酸或聚桂醇时,作为聚合物15,可以组合甲基丙烯酸氨基烷基酯共聚物、甲基丙烯酸铵基烷基酯共聚物、甲基丙烯酸共聚物、羟丙甲纤维素乙酸酯丁二酸酯或聚乙烯吡咯烷酮。更优选地,硬脂酸可以用作熔融成分13,作为聚合物15可与熔融成分13组合的聚合物为甲基丙烯酸氨基烷基酯共聚
物、甲基丙烯酸铵基烷基酯共聚物或聚乙烯吡咯烷酮。或者,可以将聚桂醇用作熔融成分13,作为聚合物15可与熔融成分13组合的聚合物为甲基丙烯酸氨基烷基酯共聚物、甲基丙烯酸铵基烷基酯共聚物、甲基丙烯酸共聚物或羟丙甲纤维素乙酸酯丁二酸酯。
30.在颗粒10中,原料药11、熔融成分13和聚合物15只要形成颗粒即可,可以是原料药11、熔融成分13和聚合物15熔化并相互混合的结构,也可以是原料药11、熔融成分13和聚合物15的一部分熔化并相互粘合的结构。
31.颗粒10优选含有原料药11作为主要成分。颗粒10中,相对于原料药11、熔融成分13及聚合物15的质量的合计,优选含有50质量%以上的原料药11。换句话说,颗粒10优选在可形成的范围内含有较少的熔融成分13和聚合物15。由此,可以有效地提高颗粒10中原料药11的含量。此外,颗粒10的颗粒粒径具有高均匀性。
32.[颗粒的制备方法]
[0033]
图2是说明根据本发明一个实施方式的制备颗粒的方法的流程图。将原料药11、熔融成分13和聚合物15混合,通过熔融造粒法熔化原料药11、熔融成分13和聚合物15并造粒,从而形成颗粒10(s101)。此时,将这些添加剂的温度加热到熔融成分13的熔点以上且聚合物15的玻璃化转变点以上的温度。考虑到熔融造粒法一般使用的温度范围,加热温度在100℃以下。优选在原料药11不变性或不发现类似物质显著增加的温度范围内进行熔融造粒。
[0034]
通过在这种温度控制下制备颗粒10,原料药11、熔融成分13和聚合物15可以粘合而制备出颗粒10。因此,可以通过熔融造粒法简便地制备颗粒10。
[0035]
[制剂]
[0036]
可以使用颗粒10制造制剂。例如,颗粒10和医药上容许的已知添加剂可以混合作为药物组合物。此外,药物组合物可以压片作为片剂。此外,可以将添加了崩解剂的药物组合物压片作为口腔崩解片。此外,药物组合物可以封装在胶囊中作为胶囊片。
[0037]
实施例
[0038]
[试验例]
[0039]
将熔融成分1g、聚合物1g混合后,在80℃下加热2小时。另外,第一熔融成分2g同样在80℃、加热2小时,用手感评价了没有混合了聚合物的熔融成分和混合有聚合物的熔融成分的粘度比较。熔融成分使用聚桂醇(nippon surfactant industries co.,ltd.)、硬脂酸(日油股份有限公司)或硬化油(freund corporation,lubriwax)。此外,聚合物使用甲基丙烯酸氨基烷基酯共聚物e(赢创公司,eudragit(注册商标)epo)、甲基丙烯酸铵基烷基酯共聚物rl(赢创公司,eudragit(注册商标)rlpo)、甲基丙烯酸共聚物l(赢创公司,eudragit(注册商标)l100-55)、羟丙甲纤维素乙酸酯丁二酸酯(信越化学工业股份有限公司,shin-etsu aqoat(注册商标)hpmc as lf)或聚乙烯吡咯烷酮(basf公司,k30)。
[0040]
评价的结果如表1所示。
[0041]
[表1]
[0042][0043]
◎
粘度显著增加,〇粘度增加,
×
无变化,-未评价。
[0044]
研究表明,当使用聚桂醇作为熔融成分时,甲基丙烯酸氨基烷基酯共聚物、甲基丙烯酸铵基烷基酯共聚物、甲基丙烯酸共聚物或羟丙甲纤维素乙酸酯丁二酸酯的粘度升高,显示出相溶性。尤其是,甲基丙烯酸共聚物对聚桂醇显示出了优异的相溶性。另外,作为熔融成分使用硬脂酸时,甲基丙烯酸氨基烷基酯共聚物、甲基丙烯酸铵基烷基酯共聚物、聚乙烯吡咯烷酮的粘度上升,显示出相溶性。尤其明确了甲基丙烯酸氨基烷基酯共聚物及聚乙烯吡咯烷酮对硬脂酸表现出优异的相溶性。另一方面,作为熔融成分使用硬化油时,任何聚合物都不显示相溶性。
[0045]
[实施例1]
[0046]
将作为原料药的磷酸西他列汀(sitagliptin phosphate)310.2g、作为熔融成分的硬脂酸(日油股份有限公司)37.5g、作为聚合物的甲基丙烯酸氨基烷基酯共聚物e(赢创公司,eudragit(注册商标)epo)37.5g放入转动造粒机(powrex corporation,mp-01)中,在转子转速400rpm、供气风量0.24l/分(min)~0.37l/分、供气温度82.9℃~89.6℃的条件下进行100分钟的造粒。此时,添加剂的温度为55.4℃~68.1℃。
[0047]
获得的颗粒的扫描型电子显微镜(sem)图像如图3所示。
[0048]
[比较例1]
[0049]
将作为原料药的磷酸西他列汀310.2g、作为溶融成份的硬化油(freund corporation,labliwax)37.5g和作为聚合物的甲基丙烯酸氨基烷基酯共聚物e(赢创公司,eudragit(注册商标)epo)37.5g投入到转动造粒机(powrex corporation,mp-01)中,在转子转速400rpm、供气风量0.24l/分~0.37l/分、供气温度82.9℃~89.6℃的条件下进行了100分钟的造粒。此时,添加剂的温度为55.4℃~68.1℃。由于硬化油与甲基丙烯酸氨基烷基酯共聚物e的相溶性不佳,造粒工序后各成分未能造粒而仍然是粉末混合物。
[0050]
将磷酸西他列汀作为原料药,硬脂酸(日油股份有限公司)作为熔融成分,甲基丙烯酸氨基烷基酯共聚物e(赢创公司,eudragit(注册商标)epo)作为聚合物,按照表2所示的添加量,放入于转动制粒机(powrex corporation,mp-01)中,并在不同的生产条件下生产颗粒。另外,使用激光衍射/散射法测定装置(beckman coulter,inc.ls 13 320)测定了制造的颗粒的粒径。
[0051]
[表2]
[0052][0053][0054]
(-)未测量。
[0055]
从实施例2~实施例4的结果来看,本发明的颗粒的原料药含量相对于颗粒总质量高达80~90质量%,比例非常高。另外,实施例2~实施例4的颗粒的粒度分布度分别为0.9~1.4,表明颗粒均匀。
[0056]
由实施例5~实施例7和比较例2的结果可知,在硬脂酸和eudragit(注册商标)epo
的组合中,在相对于颗粒总质量以86质量%的非常高的比率含有原料药的配方条件下,熔融成分和聚合物的配合比率是重要的,如果以熔融成分和聚合物的质量比计含有4∶1以上的聚合物,且含有少于2∶3的聚合物,则可以制造颗粒。
[0057]
(附图标记的说明)
[0058]
10:颗粒;11:原料药;13:熔融成分;15:聚合物。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1