用于治疗疼痛的结合TGF-α和表皮调节素的抗体的制作方法
用于治疗疼痛的结合tgf-α
和表皮调节素的抗体
1.本发明涉及结合人tgf-α和表皮调节素(epiregulin)的抗体在治疗慢性疼痛中的用途,所述慢性疼痛包括伤害性、神经性和混合性疼痛,并且特别是在治疗慢性骨关节炎(oa)疼痛,或慢性糖尿病性周围神经病变疼痛(dpnp),或慢性腰痛中的用途。
2.慢性疼痛根据机制分为不同的类别:伤害性、神经性和混合性。伤害性疼痛是由潜在或实际导致非神经组织损伤的刺激引起的。这激活了外周感觉系统中的伤害性感受器。骨关节炎(oa)引起的疼痛是躯体伤害性疼痛的典型实例。神经性疼痛是由中枢或外周神经系统的损伤或疾病引起的,导致感觉神经系统的不适应性超敏反应。糖尿病性周围神经病变(dpnp)引起的疼痛是周围神经性疼痛的典型实例。表现出伤害性疼痛和神经性疼痛特征的病症,如慢性腰痛,被归类为混合性疼痛。
3.慢性疼痛是非常普遍的病症,具有巨大的社会影响。2016年,根据国家健康访谈调查的数据,美国估计有20.4%的成年人口经历了慢性疼痛,慢性疼痛定义为在过去6个月的大多数日子或每天都会感到疼痛。据估计,8%的人口患有慢性疼痛,在过去6个月的大多数日子或每天都会限制他们的生活或工作活动。因此,慢性疼痛是保健支出的主要原因,2010年美国每年管理慢性疼痛的费用估计约为6350亿美元。尽管疾病负担和社会影响很高,但慢性疼痛的管理目前并不令人满意。单纯的非药物治疗很少足以缓解疼痛或改善功能,可用的药物治疗提供了适度的益处,并具有显著的安全性风险。目前,最常用的缓解慢性疼痛的药物是对乙酰氨基酚、非甾体抗炎药和阿片类药物。加巴喷丁类药物、其他抗惊厥药(如二氯戊酸钠(sodium divalproate)、卡巴咪嗪或拉莫三嗪)和一些抗抑郁药(如三环类药物或度洛西汀)可用于某些特定的疼痛疾病。目前的药理学药物通常表现出低水平的功效、耐受性问题和/或有害副作用。阿片类药物对急性疼痛有效,但由于滥用风险高和潜在的严重不良反应,它们对慢性疼痛的治疗选择有限。慢性疼痛对患者和社会的身体、情感和经济影响,加上缺乏有效和可耐受的治疗方案,使其成为一种严重未满足的医疗需求。
4.数据表明,egfr通路参与神经性疼痛的发病机制(kersten,c,cameronmg,laird b,s.epidermal growth factor receptor-inhibition(egfr-i)in the treatment of neuropathic pain.br j anaesth.2015;115(5):761-767)。然而,用egfr抗体或egfr酪氨酸激酶抑制剂靶向受体与胃肠道(gi)和皮肤不良反应的高发生率相关,这限制了它们在慢性疼痛的非肿瘤人群中的潜在应用。结合并激活egfr的配体包括表皮调节素(ereg)、转化生长因子α(tgfα)、表皮生长因子、肝素结合表皮样生长因子、β细胞素、双调节蛋白和epigen(schneider mr,wolf e.the epidermal growth factor receptor ligands at a glance.j cell physiol.2009;218(3):460-466)。两种egfr配体,tgfα和表皮调节素,其独特之处在于它们不能诱导受体降解,从而促进受体循环和持续的egfr途径激活(roepstorff k,grandal mv,henriksen l,knudesen sl,lerdrup m,l,willumsen bm,van deurs b.differential effects of egfr ligands on endocytic sorting of the receptor.traffic.2009;10(8):1115-1127)。
5.抗体i是一种高亲和力的人源化免疫球蛋白g4(igg4)单克隆抗体,其与人tgfα和表皮调节素的c末端区域中的关键残基结合,防止其与egfr的结合和激活,并且抗体i和制
备该抗体的方法及其制剂以及糖尿病性肾病的治疗方法公开于wo 2012/138510中。
6.对于慢性疼痛(包括伤害性、神经性和混合性疼痛)的替代治疗和或改进的治疗,特别是在骨关节炎(oa),或糖尿病性周围神经病变(dpnp),或慢性腰痛的治疗中,仍有未满足的需求。此外,对于慢性疼痛(包括伤害性、神经性和混合性疼痛)的替代治疗和或改进的治疗,特别是在骨关节炎(oa),或糖尿病性周围神经病变(dpnp),或慢性腰痛的治疗中,仍存在未满足的需求,以治疗难治性疼痛,其在本文中被定义为两种或多种先前的单一治疗和/或双重治疗的治疗方案难以治疗的疼痛。
7.本发明提供了针对tgf-α和表皮调节素的抗体,用于治疗慢性疼痛,包括伤害性、神经性和混合性疼痛,特别是治疗慢性骨关节炎(oa)疼痛,或慢性糖尿病性周围神经病变疼痛(dpnp),或慢性腰痛。此外,本发明提供了针对tgf-α和表皮调节素的抗体,用于治疗两种或多种先前的单一治疗和/或双重治疗的治疗方案难以治疗的慢性骨关节炎(oa)疼痛、慢性糖尿病性周围神经病变疼痛(dpnp),或慢性腰痛。
8.在一个实施方案中,本发明提供一种治疗有需要的受试者的慢性疼痛的方法,包括向受试者施用治疗有效量的包含轻链和重链的抗体,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr包含氨基酸序列lcdr1、lcdr2和lcdr3,和hcvr包含氨基酸序列hcdr1、hcdr2和hcdr3,其中lcdr1是seq id no:4,lcdr2是seq id no:5,lcdr3是seq id no:6,hcdr1是seq id no:1,hcdr2是seq id no:2和hcdr3是seq id no:3。
9.在一个实施方案中,本发明提供了一种治疗有需要的受试者的慢性骨关节炎疼痛的方法,包括向受试者施用治疗有效量的包含轻链和重链的抗体,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr包含氨基酸序列lcdr1、lcdr2和lcdr3,和hcvr包含氨基酸序列hcdr1、hcdr2和hcdr3,其中lcdr1是seq id no:4,lcdr2是seq id no:5,lcdr3是seq id no:6,hcdr1是seq id no:1,hcdr2是seq id no:2和hcdr3是seq id no:3。
10.在一个实施方案中,本发明提供了一种治疗有需要的受试者的慢性糖尿病性周围神经病变疼痛的方法,包括向受试者施用治疗有效量的包含轻链和重链的抗体,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr包含氨基酸序列lcdr1、lcdr2和lcdr3,和hcvr包含氨基酸序列hcdr1、hcdr2和hcdr3,其中lcdr1是seq id no:4,lcdr2是seq id no:5,lcdr3是seq id no:6,hcdr1是seq id no:1,hcdr2是seq id no:2和hcdr3是seq id no:3。
11.在一个实施方案中,本发明提供了一种治疗有需要的受试者的慢性腰痛的方法,包括向受试者施用治疗有效量的包含轻链和重链的抗体,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr包含氨基酸序列lcdr1、lcdr2和lcdr3,和hcvr包含氨基酸序列hcdr1、hcdr2和hcdr3,其中lcdr1是seq id no:4,lcdr2是seq id no:5,lcdr3是seq id no:6,hcdr1是seq id no:1,hcdr2是seq id no:2和hcdr3是seq id no:3。
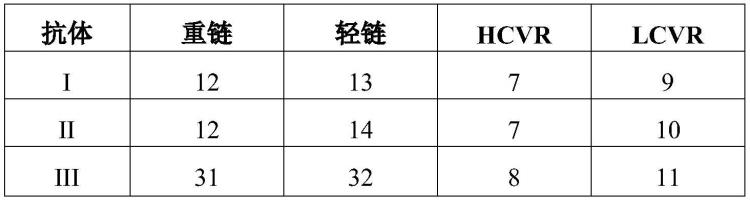
12.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,其中lcvr的氨基酸序列是seq id no:9或seq id no:10。
13.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,其中hcvr的氨基酸序列是seq id no:7。
14.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,其中lcvr的
氨基酸序列是seq id no:9和hcvr的氨基酸序列是seq id no:7。
15.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,其中轻链的氨基酸序列是seq id no:13或seq id no:14。
16.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,其中重链的氨基酸序列是seq id no:12。
17.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,包含两条轻链和两条重链,其中每条轻链的氨基酸序列是seq id no:13和每条重链的氨基酸序列是seq id no:12。
18.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,包含两条轻链和两条重链,其中每条轻链的氨基酸序列是seq id no:14和每条重链的氨基酸序列是seq id no:12。
19.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,其中抗体的剂量为750mg起始剂量,接着为每两周500mg剂量,持续患者治疗疼痛需要的时间。
20.在一个实施方案中,本发明提供了根据上述任一个实施方案的方法,其中慢性疼痛是两种或多种先前的单一治疗和/或双重治疗的治疗方案难以治疗的。
21.在一个实施方案中,本发明提供了包含轻链和重链的抗体,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr包含氨基酸序列lcdr1、lcdr2和lcdr3,和hcvr包含氨基酸序列hcdr1、hcdr2和hcdr3,其中lcdr1是seq id no:4,lcdr2是seq id no:5,lcdr3是seq id no:6,hcdr1是seq id no:1,hcdr2是seq id no:2和hcdr3是seq id no:3,用于慢性疼痛的治疗中。在一个实施方案中,本发明进一步提供了以上用途,其中慢性疼痛选自慢性骨关节炎疼痛、慢性糖尿病性神经病变疼痛和慢性腰痛。在一个实施方案中,本发明进一步提供了上述用途,其中lcvr的氨基酸序列是seq id no:9或seq id no:10。在一个实施方案中,本发明进一步提供了上述用途,其中hcvr的氨基酸序列是seq id no:7。在一个实施方案中,本发明进一步提供了上述用途,其中lcvr的氨基酸序列是seq id no:9和hcvr的氨基酸序列是seq id no:7。在一个实施方案中,本发明进一步提供了上述用途,其中轻链的氨基酸序列是seq id no:13或seq id no:14。在一个实施方案中,本发明进一步提供了上述用途,其中重链的氨基酸序列是seq id no:12。在一个实施方案中,本发明进一步提供了上述用途,包括两条轻链和两条重链,其中每条轻链的氨基酸序列是seq id no:13和每条重链的氨基酸序列是seq id no:12。在一个实施方案中,本发明进一步提供了上述用途,包括两条轻链和两条重链,其中每条轻链的氨基酸序列是seq id no:14和每条重链的氨基酸序列是seq id no:12。在一个实施方案中,本发明进一步提供了上述用途,其中慢性疼痛是慢性骨关节炎疼痛。在一个实施方案中,本发明进一步提供了上述用途,其中慢性疼痛是慢性糖尿病性周围神经病变疼痛。在一个实施方案中,本发明进一步提供了上述用途,其中慢性疼痛是慢性腰痛。在一个实施方案中,本发明进一步提供了上述用途,其中抗体的剂量为750mg起始剂量,接着为每两周500mg剂量,持续患者治疗疼痛需要的时间。在一个实施方案中,本发明进一步提供了上述用途,其中慢性疼痛是两种或多种先前的单一治疗和/或双重治疗的治疗方案难以治疗的。在一个实施方案中,本发明提供了根据上述实施方案的抗体用于制备治疗慢性疼痛的药物的用途。在一个实施方案中,本发明提供了根据上述实施方案的抗体用于制备治疗慢性疼痛的药物的用途,其中慢性疼痛选自慢
性骨关节炎疼痛、慢性糖尿病性神经病变疼痛和慢性腰痛。
22.附图简述
23.图1.抗体iii在大鼠半月板撕裂模型中的疼痛疗效。数据以平均值
±
sem表示,其中组大小为n=6。统计比较:anova和dunnett检验用于与对照igg1的比较(*p《0.001),以及tukey检验用于1和10mg/kg抗体iii之间的比较(*p《0.05)。缩写:anova=方差分析;igg1=免疫球蛋白g1;sem=平均值的标准误差。
24.图2.26周、2期、随机、双盲、安慰剂对照研究的综述,所述研究将抗体i与安慰剂进行比较。
25.如本文所使用的,慢性疼痛是指持续超过一天的疼痛,或一个月内复发数次的疼痛。慢性骨关节炎疼痛、慢性糖尿病性神经病变疼痛和慢性腰痛以及患有这些病症的患者的鉴定可以通过本领域技术人员已知的方法使用既定标准(包括本文所述的那些)来确定。如本文所用,患者是被诊断为患有需要用本文所述的抗体治疗的病症或病症的人。在其中可通过本发明方法治疗的疾病通过已建立和公认的分类是已知的那些情况下,如骨关节炎疼痛、糖尿病性神经性疼痛或腰痛,其分类可在各种来源中找到,并且国际疾病分类第10版(international classification of diseases,tenth revision(icd-10))提供了本文所述疾病的分类。本领域技术人员将认识到,对于本文所述的疾病,包括dsm-iv和icd-10中所述的那些,存在替代的命名法、分类法和分类系统,并且术语和分类系统随着医学科学的进步而发展。
26.如本文所用,骨关节炎疼痛明确包括非神经根性疼痛(非神经性疼痛)。如本文所用,神经性疼痛明确包括神经根疼痛clbp、dnp和lsr。在实施方案中,疼痛是慢性疼痛,如,例如,通过本方法治疗的骨骼肌以及神经源性的慢性疼痛。在其他实施方案中,通过本方法治疗的疼痛是内脏疼痛(如,例如,慢性前列腺炎、间质性膀胱炎(膀胱疼痛)或慢性盆腔疼痛)。
27.其他实施方案提供了治疗伤害性(nociceptive)/炎性、神经性、伤害性(nociplastic)或混合病因的疼痛的方法。在其他实施方案中,疼痛是起源于肌肉骨骼或神经性的慢性疼痛。通过本发明方法治疗的其他类型的疼痛包括手术后疼痛、类风湿性关节炎疼痛、神经性疼痛和骨关节炎疼痛。
28.示例性疼痛类型包括神经性疼痛,例如,疼痛的糖尿病性神经病变、化疗诱导的周围神经病变、腰痛、三叉神经痛、带状疱疹后神经痛、坐骨神经痛和复杂的区域疼痛综合征;炎性疼痛,如来自类风湿性关节炎、骨关节炎、颞下颌关节紊乱;pdn或cipn;内脏疼痛,如来自胰腺炎、炎性肠病、结肠炎、克罗恩病、子宫内膜异位症、盆腔疼痛和心绞痛;选自癌症疼痛、烧伤疼痛、口腔疼痛、挤压和损伤引起的疼痛、切口疼痛、骨痛、镰状细胞病疼痛、纤维肌痛和肌肉骨骼疼痛的疼痛;或来自痛觉过敏或异常性疼痛的疼痛。
29.疼痛,如本文所定义的,明确包括骨骼肌以及神经源性的慢性疼痛。“手术后疼痛”(可互换地称为“切口后疼痛”或“创伤后疼痛”)是指由外部创伤引起或导致的疼痛,所述外部创伤如切割口、穿刺、切口、撕裂或个体组织中的伤口(包括所有手术程序引起的疼痛,无论是有创的还是无创的)。如本文所用,手术后疼痛不包括在没有外部物理创伤的情况下发生(产生或起源)的疼痛。在一些实施方案中,手术后疼痛是内部或外部(包括外周)疼痛,并且伤口、切割口、创伤、撕裂或切口可能是意外地(如创伤伤口)或故意地(如手术切口)产
生。如本文所用,“疼痛”包括痛觉和疼痛感,并且可以使用疼痛评分和本领域公知的其他方法客观和主观地评估疼痛。如本文所用,手术后疼痛包括异常性疼痛(即,对正常无害刺激的反应增加)和痛觉过敏(即,对通常有害或不愉快刺激的反应增强),其转而本质上可以是热的或机械的(触觉)。在一些实施方案中,疼痛的特征在于热敏感性、机械敏感性和/或静息疼痛。在一些实施方案中,手术后疼痛包括机械引起的疼痛或静息疼痛。在其他实施方案中,手术后疼痛包括静息疼痛。疼痛可以是本领域公知的原发性或继发性疼痛。
30.如本文可交换使用的,术语“患者”、“受试者”和“个体”是指人。在某些实施方案中,患者进一步用将得益于tgf-α和表皮调节素抑制的疾病、失调或病症(例如,疼痛)来表征。
31.术语“治疗(treating)”(或“治疗(treat)”或“治疗(treatment)”)表示减缓、停止、降低或逆转症状、失调、病症或疾病的进展或严重性。
32.有效量可由本领域技术人员通过使用已知技术并通过观察在类似情况下获得的结果来确定。在确定患者的有效量时,需要考虑许多因素,包括但不限于:患者的物种;其大小、年龄和总体健康状况;所涉及的特定疾病或病症;疾病或病症的程度、涉及或严重性;个体患者的反应;所施用的特定化合物;给药模式;给药制剂的生物利用度特征;选择的剂量方案;同时用药;以及其他相关情况。
33.术语“治疗有效量”是指本发明的抗体的含量或剂量,其在以单个或多个剂量施用于患者时,提供了所需的治疗。在一些实施方案中,有效量提供了临床上显著的疼痛减轻。每周、每两周、每月或每季度胃肠外(包括,但不限于,皮下、肌内和/或静脉内)剂量可以为约0.5mg/kg至约50mg/kg。
34.每周、每两周、每月或每季度胃肠外(包括,但不限于,皮下、肌内和/或静脉内)剂量可以为约0.5mg/kg至约10mg/kg,约1mg/kg至约10mg/kg,约2mg/kg至约10mg/kg,约3mg/kg至约10mg/kg,约4mg/kg至约10mg/kg,约5mg/kg至约10mg/kg,约6mg/kg至约10mg/kg,约7mg/kg至约10mg/kg from about 8mg/kg至约10mg/kg,约1mg/kg至约8mg/kg,约2mg/kg至约8mg/kg,约3mg/kg至约8mg/kg,约4mg/kg至约8mg/kg,约5mg/kg至约8mg/kg,约6mg/kg至约8mg/kg,约1mg/kg至约6mg/kg,约2mg/kg至约6mg/kg,约3mg/kg至约6mg/kg,约4mg/kg至约6mg/kg,约5mg/kg至约6mg/kg,约1mg/kg至约5mg/kg,约2mg/kg至约5mg/kg,约3mg/kg至约5mg/kg,约4mg/kg至约5mg/kg,约1mg/kg至约4mg/kg,约2mg/kg至约4mg/kg,约3mg/kg至约4mg/kg,约3.5mg/kg至约5mg/kg,或约4mg/kg至约5mg/kg。
35.每周、每两周、每月或每季度胃肠外(包括,但不限于,皮下、肌内和/或静脉内)剂量可以为例如约50mg至约500mg,约75mg至约500mg,约100mg至约500mg,约125mg至约500mg,约250mg至约500mg,约300mg至约500mg,约350mg至约500mg,约400mg至约500mg,约450mg至约500mg,约50mg至约400mg,约75mg至约400mg,约100mg至约400mg,约125mg至约400mg,约250mg至约400mg,约300mg至约400mg,约350mg至约400mg,约50mg至约300mg,约75mg至约300mg,约100mg至约300mg,约125mg至约300mg,约150mg至约300mg,约175mg至约300mg,约200mg至约300mg,约250mg至约300mg,约50mg至约250mg,约75mg至约250mg,约100mg至约250mg,约125mg至约250mg,约150mg至约250mg,约175mg至约250mg,约200mg至约250mg,约75mg至约250mg,约50mg至约200mg,约75mg至约200mg,约100mg至约200mg,约125mg至约200mg,约150mg至约200mg,约175mg至约200mg,约50mg至约175mg,约75mg至约
175mg,约100mg至约175mg,约125mg至约175mg,或约150mg至约175mg。
36.然而,也考虑了低于或高于本文所提及剂量的剂量也,由其是考虑了本领域技术人员已知的和/或本文所述的剂量考虑事项。可以通过周期性评估监测所治疗患者的进展,并且根据需要调整剂量。
37.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr包含氨基酸序列lcdr1、lcdr2和lcdr3,和hcvr包含氨基酸序列hcdr1、hcdr2和hcdr3,其中lcdr1是seq id no:4,lcdr2是seq id no:5,lcdr3是seq id no:6,hcdr1是seq id no:1,hcdr2是seq id no:2和hcdr3是seq id no:3。
38.此外,本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr的氨基酸序列为seq id no:9或seq id no:10。
39.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中hcvr的氨基酸序列为seq id no:7。
40.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr的氨基酸序列和hcvr的氨基酸序列选自:
41.(7)lcvr是seq id no:9和hcvr是seq id no:7;和
42.(ii)lcvr是seq id no:10和hcvr是seq id no:7。
43.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr的氨基酸序列为seq id no:9和hcvr的氨基酸序列为seq id no:7。
44.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中轻链包含轻链可变区(lcvr)和重链包含重链可变区(hcvr),其中lcvr的氨基酸序列为seq id no:10和hcvr的氨基酸序列为seq id no:7。
45.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中轻链的氨基酸序列为seq id no:13或seq id no:14。
46.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中重链的氨基酸序列为seq id no:12。
47.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含轻链和重链,其中重链的氨基酸序列和轻链的氨基酸选自:
48.(i)重链是seq id no:12和轻链是seq id no:13。
49.(ii)重链是seq id no:12和轻链是seq id no:14。
50.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含两条轻链和两条重链,其中每条轻链的氨基酸序列是seq id no:13和每条重链的氨基酸序列是seq id no:12。
51.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包含两条轻链和两条重链,其中每条轻链的氨基酸序列是seq id no:14和每条重链的氨基酸序列是seq id no:
12。
52.本发明的方法中使用的抗体结合tgf-α和表皮调节素,并且包括包含本文所述的抗体和至少一种药学上可接受的载体、稀释剂或赋形剂的组合物。
53.本发明的方法中使用的抗体可以包含药物组合物,所述药物组合物包含本文所述的抗体,与至少一种药学上可接受的载体、稀释剂或赋形剂以及任选的其他治疗成分一起。
54.本发明还提供了治疗患者慢性疼痛的方法,包括将本文所述的本发明的抗体与标准护理分开、同时或按序组合施用于患者。
55.此外,本发明提供了使用本文所述的抗体的方法,用于治疗中,其中抗体与标准护理同时或按序组合施用。优选,本发明提供了使用本文所述的抗体的方法,用于慢性疼痛的治疗中,其中将抗体与标准护理同时或按序组合施用。
[0056]“抗体”的一般结构在本领域中是众所周知的。对于igg型抗体,有四条氨基酸链(两条“重”链和两条“轻”链),其通过链内和链间二硫键交联。在某些生物系统中表达时,具有未修饰的人fc序列的抗体在fc区糖基化。抗体也可以在其他位置糖基化。抗体的亚单位结构和三维构型在本领域是众所周知的。每条重链由n末端重链可变区(“hcvr”)和重链恒定区(“hccr”)组成。重链恒定区对于igg、igd和iga由三个结构域(ch1、ch2和ch3);以及对于igm和ige由4个结构域(ch1、ch2、ch3和ch4)组成。每条轻链由轻链可变区(“lcvr”)和轻链恒定区(“lcor”)组成。
[0057]
每个轻/重链对的可变区形成抗体结合位点。hcvr和lcvr区可进一步细分为高变区,称为互补决定区(“cdr”),中间散布着更保守的区域,称为框架区(“fr”)。每个hcvr和lcvr由三个cdr和四个fr组成,从氨基末端到羧基末端按以下顺序排列:fr1、cdr1、fr2、cdr2、fr3、cdr3、fr4,重链的3个cdr被称为“cdrh1、cdrh2和cdrh3”,而轻链的3个cdr被称“cdrl1、cdrl2和cdrl3”。cdr含有与抗原形成特异性相互作用的大部分残基。对每个结构域的氨基酸分配符合众所周知的惯例[例如,kabat,“sequences of proteins of immunological interest,”national institutes of health,bethesda,md.(1991)]。
[0058]
本发明中使用的抗体可以具有选自任一种免疫球蛋白类别(iga、igd、igg、igm和ige)的重链恒定区。此外,本发明中使用的抗体含有源自人igg4 fc区的fc部分,因为与其他igg亚型相比,其降低了结合补体因子的能力。
[0059]
抗体可以源自单个拷贝或克隆,包括例如任何真核、原核或噬菌体克隆。优选,本发明中使用的抗体以抗体分子的同质或基本上同质的群存在。全长抗体包括全长或基本上全长恒定区,包括fc区。这样的抗体的“抗原结合片段”是任何截短形式的全长抗体,其包含抗原结合部分并保留抗原结合能力。这样的截短形式包括例如fab片段、fab’片段或f(ab’)2片段,其包括所公开的抗体的cdr或可变区。此外,这样的截短的抗体形式可以是单链fv片段,其可以通过将编码lcvr和hcvr的dna与接头序列连接来产生。(参见,pluckthun,the pharmacology of monoclonal antibodies,vol.113,rosenburg和moore编辑,springer-verlag,new york,pp 269-315,1994)。术语“抗体”不包括这样的片段,除非另外所述。本发明中使用的抗体可以使用本领域公知的技术来生产,例如,重组技术、噬菌体展示技术、合成技术或此类技术的组合或本领域容易获知的其他技术。
[0060]
本发明使用的抗体是工程化抗体,其已经设计为具有与源自人基因组序列的框架和恒定区相同或基本上相同(基本上是人的)的人来源的框架、铰链区和恒定区。完全人框
架、铰链区和恒定区是那些人种系序列以及具有天然产生的体突变的序列和具有工程化突变的那些。本发明中使用的抗体可以包含源自完全人框架、铰链或恒定区的其中含有一个或多个氨基酸置换、缺失或添加的框架、铰链或恒定区。此外,本发明中使用的抗体是在人中基本上是非免疫原性的。
[0061]
各种不同的人框架序列可以单独或组合使用,作为用于本发明中使用的抗体的基础。优选,本发明的抗体的框架区是人源的或基本上是人的(至少95%、97%或99%人源的)。人源的框架区序列可以获自the immunoglobulin factsbook,由marie-paule lafranc,gerard lefranc,academic press 2001,isbn 012441351。
[0062]
本发明中使用的抗体的框架区系列用作“供体”可变框架区并且可以使用本领域已知的方法用于形成具有本文指定的相同cdr的其他抗体。此外,本发明中使用的抗体的框架序列可以与其他已知的人框架序列进行比较,以生成其他抗体。因此,这个信息可以用于将另一个选定的同源人框架区“回复突变”成这些位置的供体氨基酸残基。此外,任何“稀有”氨基酸可以在其他人框架中检测,使得在相关位置可以使用共有或供体氨基酸残基。
[0063]“tgf-α”或“人tgf-α”是指人tgf-α蛋白(seq id no:18)。
[0064]“表皮调节素”或“人表皮调节素”是指人表皮调节素蛋白(seq id no:33)。将met-人表皮调节素(seq id no:22)用于本文的体外实验中。关于本文所述的抗体结合或中和人表皮调节素的能力也适用于它们在体外实验中结合和中和人met-表皮调节素的能力。
[0065]
基本上按照以下所述的进行以下实施例。
实施例
[0066]
实施例1生产抗体
[0067]
可以如下制得并纯化抗体i和ii。使用最佳预定的hc:lc载体比例或编码hc(如seq id no:15)和lc(如seq id no:16或seq id no:17)两者的单一载体系统,用表达系统瞬时或稳定转染合适的宿主细胞,如hek29或cho,所述表达系统用于分泌抗体。使用许多常用技术中的任何一种纯化其中已经分泌了抗体的澄清培养基。例如,可以将培养基方便地施加于已经用相容缓冲剂(如磷酸盐缓冲盐水(ph7.4))平衡的蛋白a或g柱。洗涤柱以除去非特异性结合组分。例如,通过ph梯度(如0.1m磷酸钠缓冲液ph6.8至0.1m柠檬酸钠缓冲液ph2.5)洗脱结合的抗体。如通过sds-page检测抗体级分,然后合并。更多纯化是任选的,这取决于所计划的用途。抗体可以是使用常规技术浓缩的和/或无菌过滤的。可以通过常规技术,包括尺寸排阻、疏水性相互作用、离子交换或羟磷灰石色谱,来有效地除去可溶性聚集物和多聚体。这些色谱步骤后的抗体纯度大于99%。可以将产物立即在-70℃下冷冻或冻干。以下提供了这些抗体的氨基酸序列。
[0068]
seq id no
[0069]
[0070][0071]
制剂:
[0072]
以玻璃小瓶中的冻干粉形式提供抗体i的药品,以用于临床试验。小瓶内容物可以用无菌0.9%氯化钠,美国药典(usp)重建/稀释。冻干药品抗体i由抗体i以及赋形剂柠檬酸钠、柠檬酸、聚山梨酸酯80和蔗糖组成。小瓶制成递送75mg抗体i。用3.2ml无菌0.9%氯化钠,usp重建/稀释小瓶内容物,产生ph6.0的由25mg/ml抗体i组成的澄清溶液。
[0073]
实施例2:针对抗体i通过表面等离子共振(biacore)测量亲和力结合
[0074]
将biacore t2000仪器(ab,upsala,sweden)、试剂和biacore t2000评估软件4.1版用于表面等离子共振分析。使用制造商的edc/nhs胺偶联方法制备cm5芯片。通过以10μl/min的速度注射edc/nhs的1:1混合物7分钟,激活所有四个流动池的表面。山羊抗人fcγ特异性抗体在10mm乙酸盐、ph 4.0缓冲液中稀释至50μg/ml,并以10μl/min的流速注射7分钟,在所有四个流动池上固定约10000ru。以10μl/min的速度注入乙醇胺7分钟,封闭未反应的位点。以30μl/min的速度注射3
×
20秒ph1.5的甘氨酸,以去除非共价结合的蛋白质。运行缓冲液为hbs-ep[10mm hepes,150mm氯化钠,3mm edta,0.005%聚山梨醇酯20]。
[0075]
在研究1中,将抗体i在运行缓冲液中稀释至50μg/ml,并在流动池2中捕获大约400-600ru。将人tgfα(seq id no:18)、大鼠tgfβ(seq id no:20)、met-人表皮调节素(seq id no:22)和食蟹猴表皮调节素(seq id no:24)在运行缓冲中从100μg/ml稀释至200nm,然后在运行缓冲中将其连续两倍稀释至6.25nm。将小鼠表皮调节素(seq id no:23)在运行缓冲液中从100μg/ml稀释至4μm,然后在运行缓冲中连续两倍稀释至125nm。每种配体浓度一式两份以30μl/min注入300秒,然后进入解离阶段。人和大鼠tgf-α的解离阶段为1800秒,人和食蟹猴表皮调节素为1200秒,小鼠表皮调节素为120秒。通过在所有流动池中以30μl/min的速度注射ph1.5的10mm甘氨酸3
×
20秒进行再生。
[0076]
在研究2中,将抗体iii在运行缓冲液中稀释至100μg/ml,并在流动池2中捕获约400-600ru。将小鼠tgf-α(seq id no:19)在运行缓冲中从100μg/ml稀释至200nm,然后在运行缓冲溶液中连续两倍稀释至6.25nm。将小鼠表皮调节素(seq id no:23)在运行缓冲液中从100μg/ml稀释至4μm,然后在运行缓冲中连续两倍稀释至125nm。每种配体浓度一式两份以30μl/min注入300秒,然后进入解离阶段。小鼠tgf-α的解离阶段为1800秒,而小鼠表皮调节素为120秒。通过在所有流动池中以30μl/min的速度注射ph1.5的10mm甘氨酸30秒来进行再生。
[0077]
减去参考的数据收集为fc2-fc1。测量值在25℃下获得。使用“1:1(langmuir)结合”结合模型评估每个配体的结合速率(k
on
)和解离速率(k
off
)。根据以下关系式从结合动力学计算亲和力(kd):kd=k
off
/k
on
。
[0078]
表1
[0079]
抗体i的结合参数
[0080][0081]
表2
[0082]
抗体iii的结合参数
[0083][0084]
因此,抗体i特异性结合tgfα和表皮调节素并非常弱地结合epigen。通过表面等离子共振(spr)测量了抗体i与来自不同物种的tgfα、表皮调节素和epigen的表观结合动力学和亲和力。
[0085]
来自不同物种的转化生长因子α、表皮调节素和epigen产生了浓度依赖性的与抗体i的结合应答,抗体i对人和大鼠tgfα具有强亲和力(在25℃下kd分别为97.6
±
20.6pm和70.5
±
19.4pm)。食蟹猴和小鼠tgfα的结合动力学没有分开测量,因为它们分别与人和大鼠tgfα为100%相同。兔tgfα的结合动力学没有分开测量,因为针对抗体i,兔和人tgfα中表位是相同的。抗体i对人和食蟹猴表皮调节素显示出强亲和力,并且对小鼠表皮调节素显示出亲和力(在25℃下kd分别为1.29
±
0.03nm、1.05
±
0.09nm和342
±
136nm)。大鼠表皮调节素的结合动力学没有分开测量,因为针对抗体i,大鼠和小鼠表皮调节素表位是相同的。抗体i对人和小鼠epigen显示出非常弱的亲和力(在25℃下kd分别为》2μm和493
±
205nm)。
[0086]
因为抗体i是人源化的单克隆抗体,由于动物对化合物产生免疫应答的可能性,其在药理学动物模型中的应用受到限制。因此,使用小鼠单克隆抗体(抗体iii)在oa疼痛的动物模型中测定中和tgfα和表皮调节素的效果。还通过spr测量了抗体iii与小鼠tgfα、表皮调节素和epigen的结合动力学。抗体iii对tgfα显示出强亲和力,并且对表皮调节素和epigen显示出弱亲和力,在25℃下相关的kd分别为38.0
±
13.6pm、215
±
15nm和365
±
39nm。
[0087]
实施例3:egf靶配体在人结肠癌细胞系ht-29中的内化
[0088]
alexa488与抗体的缀合
[0089]
根据制造商的方案,将alexa488与抗体i和对照igg缀合。将蛋白质在pbs
中稀释至2mg/ml。向0.5ml的这种2mg/ml溶液中,加入50μl 1m碳酸氢钠ph9。然后将蛋白质溶液转移至染料小瓶中并在室温下搅拌1小时。使用包括在标记试剂盒中的bio-rad biogel p-30树脂纯化标记的蛋白质。
[0090]
体外内化测定
[0091]
在研究1中,在96孔板的每个孔中接种10,000个ht-29细胞(已知表达tgf-α和表皮调节素的结肠腺癌细胞系),并允许在完全培养基[dulbecco改良eagle培养基/f12(ham)培养基(1:1)(“dmem/f12”),含有l-谷氨酰胺、10%热灭活胎牛血清(“fbs”)、1
×
抗生素和2.438g/l碳酸氢钠]中孵育过夜。第二天,用含有0.1%bsa的pbs洗涤细胞,然后在37℃的组织培养箱中用含有0.1%bsa的pbs中的缀合alexa488的抗体i或对照igg在0至88ug/ml的浓度范围下孵育2小时。孵育期后,将细胞在含有0.1%bsa的pbs中洗涤数次,然后用4%甲醛固定用于分析。内化的定量如下:用cellomics arrayscan vti(thermo scientific)收集500个细胞/孔。图像分析是通过系统的“隔室分析”生物应用进行的。用hoechst染色(蓝色)鉴定细胞核。设置两个目标区域(roi)以收集来自从掩蔽图像获得的胞内斑点(红色)和总绿色荧光(红色和蓝色)的荧光信号。计算来自每个斑点和细胞的数量、面积和荧光强度。选择胞内斑点(红色)的平均斑点总强度来测量抗体i诱导的内化。
[0092]
在研究2中,按照之前所述制备了10,000个ht-29细胞,并且将含有0.1%bsa的pbs中的缀合alexa488的抗体i或对照igg以40ug/ml加入细胞中。将细胞在37℃的组织培养箱中孵育0-120分钟的不同时间范围,随后用含有0.1%bsa的pbs洗涤数次并用4%甲醛固定用于分析。基本上按照之前所述的进行信号的定量。
[0093]
表3a
[0094]
研究1-荧光的平均环斑总强度
[0095][0096]
表3b
[0097]
研究1-荧光的平均环斑总强度
[0098][0099]
[0100]
来自研究1的成像分析的结果确定了荧光信号内化至细胞中并且是抗体i剂量依赖性的,但对照igg没有(表3a和表3b)。
[0101]
表4
[0102]
研究2-荧光的平均环斑总强度
[0103][0104]
来自研究2的结果证明了抗体i快速内化并在加入细胞后2小时内完成内化(表4)。抗体i以时间依赖性方式在体外诱导靶标在ht-29细胞上的内化(表4)。以上结果表明抗体i结合膜结合的配体并促进它们以剂量依赖性和时间依赖性方式内化,抗体i(并推测配体)在37℃下孵育的2小时内完成内化。
[0105]
实施例4:成肌纤维细胞系中中和egfr配体刺激的细胞增殖的测量
[0106]
将克隆小鼠成肌纤维细胞系(“mfc7”)用于测试本发明的抗体阻断egfr配体的增殖活性的能力。可以激活egfr的七种配体是tgf-α(tgfa)、表皮调节素(ereg)、egf、肝素结合egf(hb-egf)、epigen(epgn)、双调蛋白(areg)和β细胞素(btc)。egfr配体共享结构基序、egf样结构域,特征在于三个分子内二硫键,其通过六个相似间隔的共有半胱氨酸残基形成。通过溴脱氧尿苷(“brdu”)掺入测定增殖活性,并根据制造商的说明用比色brdu elisa试剂盒测量。
[0107]
首先,将2000个mfc7细胞/孔铺板在组织培养处理的96孔微孔板中的0.1ml dulbecco改良eagle培养基/f12(ham)培养基(1:1)(“dmem/f12”)中,其含有l-谷氨酰胺、10%热灭活fbs、1
×
抗生素和2.438g/l碳酸氢钠。使细胞附着6小时,然后除去培养基并用0.1ml含有0.1%bsa的无血清dmem/f12代替,用于血清饥饿过夜。第二天,用含有0.1%bsa的无血清培养基在96孔聚丙烯板中进行egfr配体的连续稀释,体积为0.12ml/孔,浓度范围为0.001至3000ng/ml。稀释后,从血清饥饿的细胞除去培养基,然后用egfr配体刺激24小时。刺激后,用brdu脉冲细胞4hr,然后根据制造商的说明用比色brdu elisa试剂盒分析。
[0108]
在测试抗体i对egfr配体的特异性时,在96孔聚丙烯板中以0.06ml/孔的体积从3000nm至0.059nm的浓度范围2x或3x连续稀释抗体。抗体连续稀释后,每孔加入0.06ml egfr配体。然后将平板在37℃的加湿组织培养箱中培养30分钟。孵育后,每孔将0.1ml溶液转移到细胞中。将细胞刺激24小时。刺激后,用brdu脉冲细胞4小时,然后用比色brdu elisa试剂盒分析。在spectramax 190平板读取器(molecular devices)上生成吸光度值(450nm-690nm),并分析数据。
[0109]
表5
[0110]
mfc7测定
[0111][0112][0113]
在测定中发现,除epigen和双调蛋白外,小鼠表皮调节素和大鼠tgf-α以及所有人egfr配体都是细胞增殖的有效刺激剂(表5)。抗体i和抗体iii对人和大鼠tgf-α和人表皮调节素活性具有高亲和力(表5)。
[0114]
表5总结了对测试的egfr配体计算的ec50值和这些配体的抗体的绝对ic50值。对抗体i计算的平均ic50,对人tgf-α为0.46
±
0.03nm,对人表皮调节素为3.15
±
1.04nm。对抗体iii计算的ic50平均值,对人tgf-α为0.52+0.04nm,对人表皮调节素为1.12+0.36nm。对抗体iii计算的平均ic50值,对大鼠tgf-α为0.13
±
0.01nm,对小鼠表皮调节素为214
±
49nm。因此,抗体i和抗体iii具有高亲和力,并且具有针对人tgf-α和人表皮调节素的完全中和活性。
[0115]
总之,表皮生长因子受体配体是许多谱系细胞(包括成纤维细胞)的有效促有丝分裂因子。为了确定抗体i的中和效力和特异性,在体外评估了其对成肌纤维细胞增殖的影响。使用单个重组配体来刺激成肌纤维细胞的增殖,并测定其相对50%的有效浓度(ec
50
)。随后,在测定中评估单个配体的亚最大浓度和不同浓度的抗体i,以定量特定配体的抑制活性。抗体i对人、小鼠、大鼠和食蟹猴tgf-α(50%抑制浓度[ic
50
]值《0.5nm)和对人和食蟹猴表皮调节素(ic
50
值《3.5nm)具有有效的中和活性。抗体i对小鼠和大鼠表皮调节素(ic
50
值《350nm)以及人、食蟹猴和大鼠epigen(ic
50
值《850nm)具有较弱的中和活性,并且对测试的其他egfr配体没有可测量的活性(ic
50
》2000nm)。
[0116]
以类似的方式,在成肌纤维细胞增殖试验中评估了小鼠抗体iii的中和活性。抗体iii也有效地抑制人和大鼠tgfα和人表皮调节素(ic
50
值分别为0.521
±
0.037nm、0.131
±
0.012nm和1.12
±
0.36nm),且对小鼠表皮调节素有活性(ic
50
值为214
±
49nm)。对其他测试
的小鼠egfr配体没有可测量的活性。
[0117]
因此,抗体i是一种高亲和力的人源化免疫球蛋白g4(igg4)单克隆抗体,它与人tgfα和表皮调节素的c-末端区域的关键残基结合,防止它们与egfr结合和egfr的激活。抗体i的结合导致tgfα和表皮调节素的膜结合形式内化,并中和成熟(可溶性)配体活性。临床数据显示了健康受试者在单次剂量高达750mg后对抗体i耐受性良好,在3个月内每3周静脉注射(iv)多次剂量高达750mg后,中重度糖尿病性肾病(dn)患者具有可接受的耐受性谱。抗体i的安全性谱与egfr抗体或egfr酪氨酸激酶抑制剂的不同,其皮肤和gi不良事件(aes)发生率低。输注抗体i后,总表皮调节素浓度的剂量/浓度依赖性增加表明其在体内与表皮调节素结合。抗体i的分子、药理学和临床特性支持这样一个概念,即这种抗体具有活性、可用性、安全性和耐受性,能够有效治疗对tgfα和表皮调节素的抑制都有反应的疾病和病症。
[0118]
本公开提供了抗体i可用于治疗慢性疼痛,包括伤害性、神经性和混合性疼痛,特别是治疗骨关节炎(oa),或糖尿病性周围神经病变(dpnp),或慢性腰痛。此外,本公开提供了抗体可用于治疗两种或更多种先前单一治疗和/或双重治疗方案难以治疗的治疗骨关节炎(oa),或糖尿病性周围神经病变(dpnp),或慢性腰痛。与抗体i共享相同cdr的抗体iii在骨关节炎疼痛的临床前模型中进行了测试,发现其在如下所述的这种模型中具有治疗疼痛的功效。这支持抗体i可用于治疗本文所述的慢性疼痛障碍的概念。
[0119]
实施例5:抗tgfα抗体抗体iii在大鼠的骨关节炎性膝关节疼痛的半月板撕裂模型中的体内功效
[0120]
oa是一种慢性的、使人衰弱的关节病,导致受影响的关节疼痛。在这项研究中,测试了抗体iii在oa大鼠mt模型中防止疾病进展和减少骨关节炎样膝关节疼痛的能力。mt是一种充分描述的oa模型,其中通过内侧副韧带和内侧半月板相互作用导致膝关节手术失稳后发生关节破坏和疼痛。通过组织学评估oa疾病进展。疼痛是通过同一动物的具有诱发的oa的手术膝和未手术对侧膝之间的负重差异来测量的。与对照igg1抗体相比,1和10mg/kg的抗体iii治疗对oa疾病进展没有影响,但以剂量依赖的方式显示出统计学上显著的疼痛减轻。
[0121]
对于这项研究,使用了48只雄性刘易斯大鼠(harlan,indianapolis,in),年龄28至29周,体重345至430克。每笼3只大鼠一组圈养,在12小时的光照/12小时的黑暗循环中保持恒定温度。除了数据收集期间,动物可以随时自由获取食物和水。大鼠按体重随机分为3组,每组16只,通过手术切除右膝关节内侧副韧带和半月板,在大鼠中诱发oa。未对左膝关节进行手术切片。大鼠被给予sc剂量的10mg/kg对照小鼠igg1抗体或1或10mg/kg抗体iii。给药开始于手术当天,即就在手术前,大鼠每周给药一次,直到研究结束。剂量体积为2ml/kg,每周通过皮下注射给药一次,直到术后4周研究结束。最后一剂是在尸检前1周给药,尸检发生在手术后4周。尸检时,采集左右膝关节,用10%福尔马林锌固定,然后送往bolderbiopath进行包埋、切片、组织学染色,并由董事会认证的兽医病理学家进行评分。为了评估这个模型中的疼痛,从每个治疗组中随机选择6只大鼠,并在研究的第24天使用无能力测试(静态差异负重)评估疼痛。这测量了手术膝和未手术膝之间的后爪负重差异。对于这项研究,报告的值是3次单独测量的平均值,每只大鼠在1秒内进行测量。操作员对所选大鼠的治疗组是不知情的。
[0122]
疼痛数据以具有标准误差(
±
sem)的平均值表示。数据通过单向均值分析进行评
估。使用dunnett检验对各组进行比较,同时将tukey-kramer hsd检验与jmp统计分析程序(sas institute inc.,nc)用于进行配对比较。如果p值小于.05(p《0.05),则认为差异是显著的。
[0123]
对于组织学数据,bolderbiopath进行了分析。使用student’s t检验或mann-whitney u检验(非参数)分析数据。如果合适,通过单因素方差分析(anova)或kruskal-wallis检验(非参数)以及检验后的适当多重比较,进一步分析所有组的数据。如果p值小于.05(p《0.05),则认为差异是显著的。
[0124]
这个研究的膝关节组织学评分表明了与对照igg1相比,用抗体iii(1或10mg/kg)治疗不会显著影响大鼠内侧半月板撕裂引起的oa损伤。然而,与对照igg1相比,在1和10mg/kg剂量下,抗体iii显著减轻了疼痛(dunnett试验p<.001;(错误!未找到参考来源)。10mg/kg剂量的抗体iii也与1mg/kg剂量显著不同(p<.05,tukey kramer honest显著差异检验)。数据提供了令人惊讶和意外的证据,表明抗体iii在减轻疼痛方面有效,这是通过大鼠半月板撕裂模型中的负重差异来衡量的。
[0125]
在人类中的作用
[0126]
这部分中呈现的临床试验数据是根据良好临床实践(gcp)的原则生成的。已经完成了两项临床试验,其中93名受试者迄今已暴露于抗体i。42名健康受试者在研究i5v-mc-tgaa(tgaa)中接受单剂量(iv或sc)。在研究i5v-mc-tgab(tgab)中,51名患者接受了多次iv输注。表6总结了这些研究的设计。
[0127]
表6.临床药理学研究的列表
[0128][0129]
缩写:ada=抗药物抗体;ae=不良事件;dlco=扩散能力测量;iv=静脉内;n=受试者数量;sc=皮下;研究tgaa=研究i5v-mc-tgaa;研究tgab=研究i5v-mc-tgab。
[0130]
实施例6临床药理学
[0131]
药物动力学
[0132]
单次和多次iv给药后,抗体i呈现出非线性药物动力学,表明靶介导的药物处置(表6.1、6.2和6.3)。在低剂量下,抗体i的表观血浆清除率明显更大,具有相对短的t
1/2
。随着抗体i的剂量增加,抗体i的t
1/2
达到432小时(18天),与通常对于靶向膜结合的蛋白质的单克隆抗体观察到的相一致。在dn患者中,对于750mg iv输注,抗体i平均暴露(从时间零至时间t的auc,其中t是具有可测量浓度的最后一个时间点[auc
0-tlast
])从单剂后的2250μg*天/ml增加至第五剂后的4880μg*天/ml(表6),导致每3周施用时大约2倍的累积。单次iv输注750mg后,健康受试者和dn患者中的最大浓度(c
max
)和暴露(auc
0-tlast
)是相当的(健康受试者和dn患者中分别地平均c
max
为265μg/ml和368μg/ml;平均auc
0-tlast
为3040μg
·
天/ml和2250μg
·
天/ml)。群pk建模显示出抗体i的药物动力学在健康受试者和dn患者中相似。
[0133]
sc剂量给药后,计算的抗体i的生物利用率为38%;然而,评估的剂量(50mg)位于药物动力学为非线性的剂量范围中。因此,临床相关线性pk范围中的生物利用率可能更高。
[0134]
表6.1.tgaa研究中健康志愿者单次iv和sc给药抗体i后非隔室(noncompartmental)抗体i药代动力学参数汇总
[0135]
几何平均(cv%几何平均)
[0136][0137]
缩写:auc
0-tlast
=从时间零到时间t的浓度-时间曲线下面积,其中t是具有可测量
浓度的最后一个时间点;auc
0-∞
=从时间零到无穷大的浓度-时间曲线下面积;cl=药物的全身清除率,即iv给药的绝对清除率和sc给药的表观清除率(cl/f);c
max
=最大血清浓度;cv=变异系数;f=生物利用率;iv=静脉内;n=剂量组中的受试者数量;na=不适用;nc=不可计算;sc=皮下;研究tgaa=研究i5v-mc-tgaa;t
max
=达到最大浓度的时间;t
1/2
=消除半衰期;v
ss
=稳定状态下的分配体积。
[0138]a这些报告值中包括5名受试者的数据。
[0139]bf=(抗体i 50mg sc平均auc
0-∞
/抗体i 50mg iv平均auc
0-∞
)*100
[0140]c中值(范围)。
[0141]
来源:i5v-mc-tgaa表14.2.1.2。衍生抗体i药代动力学参数汇总。
[0142]
表6.2.tgab研究中糖尿病性肾病患者单次iv给药抗体i后非隔室抗体i药代动力学参数汇总(a部分)
[0143][0144]
缩写:auc
0-tlast
=从时间零到时间t的浓度-时间曲线下面积,其中t是具有可测量浓度的最后一个时间点;c
max
=最大血清浓度;iv=静脉内;n=剂量组中的受试者数量;研究tgab=研究i5v-mc-tgab;t
max
=达到最大浓度的时间。a中值(范围)。
[0145]
表6.3.tgab研究中糖尿病性肾病患者5q3w iv给药抗体i后非隔室抗体i药代动力学参数汇总(b部分)
[0146]
[0147]
缩写:auc
0-tlast
=从时间零到时间t的浓度-时间曲线下面积,其中t是具有可测量浓度的最后一个时间点;aucτ=一剂给药间隔期间的浓度时间曲线下面积;c
max
=最大血清浓度;iv=静脉内;n=剂量组中的受试者数量;q3w=每3周1次;研究tgab=研究i5v-mc-tgab;t
max
=达到最大浓度的时间。
[0148]a中值(范围)。
[0149]bn=4。
[0150]
药效学
[0151]
通过测量总表皮调节素和总tgfα浓度的靶标结合(target engagement):
[0152]
临床方案中使用了药物耐受性表皮调节素和tgfα测定,以测量总表皮调节素浓度(总表皮调节素或tgfα=抗体i结合的表皮调节素或tgfα+游离表皮调节素或者tgfα)。施用抗体i时,前提是表皮调节素/tgfα将与抗体i结合,可与egfr相互作用的游离表皮调节素/tgfα的量将减少。预计抗体i结合的表皮调节素tgfα的清除率低于游离表皮调节素/tgfα,从而导致施用抗体i后总表皮调节素/tgfα浓度增加。
[0153]
施用抗体i后,健康受试者和dn患者的血清总表皮调节素水平以剂量依赖性增加。单剂量抗体i施用后1至4周内,总血清表皮调节素水平通常达到峰值,并在随后的给药后继续累积。这与血清部分(compartment)中总表皮调节素的羁留,以及清除率降低且因此其血清浓度的逐渐累积相一致。这与表皮调节素结合靶标相一致。
[0154]
健康受试者单剂量iv施用抗体i后,血清tgfα水平似乎没有变化;在dn患者中施用多剂后,血清总tgfα浓度呈剂量依赖性增加。然而,血清总tgfα的增加幅度小于表皮调节素的,并且变化更大。
[0155]
总血清表皮调节素和tgfα的数据表明,抗体i与表皮调节素和tgfα两者结合,并支持临床前数据,即抗体i可以在人体内与表皮调节素和tgf-α两者结合。
[0156]
临床研究中的安全性数据汇总
[0157]
研究tgaa:这个研究中未发生与抗体i给药相关的严重不良事件(sae)。没有受试者因被认为与抗体i相关的ae而退出或停止治疗。在接受抗体i后,至少有2名(或≥5%)受试者报告了所有因果关系的治疗突发的aes(teae),并且比接受安慰剂的受试者更频繁的报告包括鼻炎、打喷嚏、背痛、咳嗽、感觉热、感觉异常和鼻漏。在随机接受抗体i的2名以上的受试者中,未观察到与研究药物相关的teae。一名受试者在给药1mg抗体i后12天出现中度严重的弥漫性瘙痒性斑丘疹。皮疹持续10天并自行消退。研究者认为皮疹与研究药物给药有关,并且与泛egfr抑制剂给药通常相关的皮疹一致。血压、心率、qtc、临床实验室测试或肺弥散能力均无临床显著异常。
[0158]
研究tgab:
[0159]
在a部分中,没有患者出现导致研究药物退出、sae或研究期间死亡的teae。接受安慰剂治疗的3名患者中有1名、接受10mg抗体i治疗的4名患者中的3名、接受100mg抗体i的4名患者中的4名和接受750mg抗体i治疗的4名患者中的2名均出现了治疗突发的aes。每个治疗组中有一名患者至少出现1次与药物相关的teae。
[0160]
在b部分中,1名接受安慰剂的患者在研究期间死亡。经历至少1次sae的患者人数包括:接受安慰剂的6名患者中的1名(16.7%)、50mg组的14名患者中的3名(21.4%)、250mg组的13名患者中的6名(46.2%)和750mg组的12名患者中的1名(8.3%)。在b部分中接受抗
体i的患者中,共有25.6%的患者报告了sae。研究中观察到的sae均未被判断为与研究药物定量给药有关,且无论治疗如何,均未有1名以上患者出现sae。
[0161]
经历了导致研究药物退出的teae的患者人数包括:接受安慰剂的6名患者中的1名(16.7%),50mg组14名患者中的2名(14.3%),250mg组13名患者中的3名(23.1%),750mg组12名患者中的1名(8.3%)。除了一个例外,所有导致研究药物退出的teae都被认为与研究药物无关。导致研究药物退出的1例相关teae是接受750mg抗体i的患者全身瘙痒。
[0162]
抗体i治疗患者在b部分中最常见的5种teae为腹泻、水肿加重、头痛、高血压恶化和恶心(各10.3%)。接受安慰剂的患者最常见的两种teae是头痛和慢性肾病(各33.3%)。没有证据表明teae的频率和抗体i存在剂量关系。研究tgab b部分中的治疗突发的aes(无论因果关系如何)总结在表6.4中。b部分中没有超过1名患者报告与药物相关的teae。随着时间从基线的生命体征值或实验室值没有一致的变化。
[0163]
表6.4.按所有抗体i优选期限的下降频率列出的治疗突发不良事件汇总
–
b部分(安全人口)
[0164]
[0165][0166]
注:使用medra版本17.0将ae编码。teae是指在治疗基线后的任何时间发生或恶化的任何不良医疗事件(即,在第一剂研究药物后发病),且不一定与该治疗具有因果关系。
[0167]
缩写:ae=不良事件;meddra=监管活动医学词典;n=剂量组中的患者数量;teae=治疗突发的ae。
[0168]
实施例7:评估抗体i治疗骨关节炎的随机安慰剂对照2期临床试验。
[0169]
这个研究的目的是为抗体i缓解骨关节炎(oa)引起的膝关节疼痛提供人类临床证据。将收集数据以评估这个研究人群中抗体i的安全性和耐受性。还将探讨药代动力学(pk)特性、药效学(pd)效应和免疫原性概况。来自这个概念验证研究的全部数据将评估与抗体i相关的益处和风险,并为抗体i的临床开发提供信息。
[0170]
整体设计:
[0171]
这是一项26周、2期、随机、双盲、安慰剂对照研究,其将在膝oa患者中比较抗体i与安慰剂。这是一项随机、研究者和参与者盲、安慰剂对照的2期临床试验。大约125名参与者将被随机分配到研究干预组(84名抗体i和41名安慰剂)。这项26周的研究包括8周的双盲治疗期和18周的随访期。该研究设计如图2所示。
[0172]
研究干预将通过不知情人员在大约1小时内缓慢静脉(iv)输注进行。如果观察到输注反应,则可根据需要降低输注速率。剂量为750mg起始剂量,然后每2周静脉注射500mg,共4剂。给药制剂是用无菌水0.9%氯化钠溶液配制的冻干粉。参与者将每2周接受一次静脉输液,共输4次。每次输注完成后,将对参与者进行至少4小时的监测。
[0173]
在患者就诊时,完成所有治疗后样本收集和安全性监测,并指示参与者在就诊解除前继续完成研究限制和数字评分量表(nrs)日记条目。根据抗体i的长pk半衰期和潜在的持续靶向结合,将在最后一剂后长达6周内收集疗效数据。将在最后给药干预后长达20周内收集安全性、药代动力学(pk)、药效学(pd)和免疫原性样本,以表征安全性和临床免疫原性谱。如果参与者已完成所有要求的研究阶段,包括最后一个计划的程序,则认为其已完成本研究。
[0174]
目标和终点:
[0175]
主要和次要目标和终点如下所述。
[0181]
情绪功能评估:eq-5d-5l健康状况问卷用于所有疾病状态。eq-5d-5l是解决生活质量问题的最受欢迎的患者完成的工具之一(buchholz i,janssen mf,kohlmann t,feng ys。a systematic review of studies comparing the measurement properties of the three-level and five-level versions of the eq-5d.pharmacoeconomics.2018;36(6):645-661.)。它是一个描述系统,其包括5个维度:行动、自我照顾、日常活动、疼痛/不适和焦虑/抑郁。参与者被要求“勾选一个最能描述你今天健康状况的选项”,从每个维度下提供的5个选项中进行选择。5个维度上的得分可以作为健康概况显示或转换为单个汇总索引编号。eq-5d-5l还包括eq vas,它在0到100的垂直vas上记录参与者的自评健康状况:0=你能想象到的最差健康状况,100=你能想到的最佳健康状况。eq-5d-5l版本中使用的工具是一个简短、可靠、经过验证、易于完成的量表,具有卓越的重新测试可靠性,以解决与一些疾病引起的疼痛相关的生活质量。
[0182]
睡眠质量:睡眠障碍是疼痛研究中的一个重要问题。在各种可用的工具中,医学结果研究(mos)睡眠量表为睡眠障碍提供了一个独特的、经心理学验证的分数。这一量表包含了过去一周的12个问题。参与者报告分为6个等级的每种睡眠症状或问题出现的频率,等级从“所有时间”到“没有时间”。关于入睡时间和睡眠量的的问题被报告为每晚平均睡眠小时数。该量表管理负担低,已用于不同的疼痛研究,并已在神经性疼痛患者中得到验证。
[0183]
安全性评估:根据典型实践确定安全性评估的计划时间点,并且包括体检、生命体征和体重测量、12导联ecg、临床实验室测试、肝脏安全性监测、c-ssrs和自发报告的aes。
[0184]
疗效评估:
[0185]
[0186][0187]
在表征慢性疼痛时,使用一组核心评估和领域,其是:疼痛、身体机能、情绪功能、参与者总体改善评分、不良事件(aes)和参与者处置。选择nrs作为主要终点是基于其已证明的检测疼痛变化的能力及其在研究疾病状态中的常用性。
[0188]
western ontario and mcmaster universities骨关节炎指数:western ontario and mcmaster universities骨关节炎索引(bellamy n.the womac knee and hip osteoarthritis indices:development,validation,globalization and influence on the development of the auscan hand osteoarthritis indices.clin exp rheumatol.2005;23(suppl.39):148-153)是经验证的工具,广泛用于评估治疗骨关节炎疼痛的药物反应。womac lk3.1版根据活动时间表进行管理。
[0189]
该表描述了24个问题的womac和子量表。
[0190]
维度问题数量疼痛5僵硬2身体机能17
[0191]
参与者将使用0至4likert量表记录每个问题的回答:0=无疼痛,4=极度疼痛。每个子量表的分数将通过对每个参与者在每个时间点的各个子量表中的问题分数进行相加来计算。每个子量表的可能得分范围为“疼痛=0至20”、“僵硬=0至8”和“身体机能=0至68”。将进行贝叶斯纵向混合模型重复测量分析(mmrm),以评估womac疼痛子量表和身体机能子量表从基线到每次基线后就诊的变化。每个治疗组中达到预先指定的二元疗效结果的参与者比例将在每个基线后时间点进行估计,并用于比较治疗组。估计值将通过拟合包括所有基线后观测值的贝叶斯纵向模型来提供。预先指定的二元疗效结果包括isa中如下的参与者比例:如通过womac疼痛子量表测量的从基线减轻大于30%、50%和70%,以及如通过womac身体功能子量表测量的从基线减轻大于30%、50%和70%。该模型将包括上述连续疗效分析模型中描述的分类和连续协变量,除了不使用基线和就诊的交互作用。统计分析计划中可考虑并规定其他模型条目。将为每个治疗组提供womac疼痛和身体机能子量表从基线到终点变化百分比的累积分布函数。
[0192]
活动时间表(soa):
[0193]
该活动时间表显示了抗体i研究的某些就诊和程序。“主方案”是指在给定疾病状态或多种疾病状态下指导若干潜在研究的方案设置,并且干预特定附录是指与研究中的给定干预相关的方案部分。
[0194]
[0195][0196]
[0197][0198]
缩写:dsa=疾病状态附录;ed=提前终止;v=就诊;womac=western ontario and mcmaster university骨关节炎指数。
[0199]a如果减少了参与者的负担,则可在随机化前的其他时间点进行筛选评估。
[0200]b站点确定参与者当前服用的每种疼痛药物的半衰期,以便安排第2次就诊。由于需要7天pdep,就诊2安排在不早于就诊3随机化前7天。
[0201]c疼痛药物的5个半衰期清除(washout)期必须在pdep之前,使得大多数参与者至少10天。
[0202]
研究人群:
[0203]
如果基于患者报告或病史有每日疼痛史,则男性和女性参与者有资格纳入研究。
[0204]
纳入标准:
[0205]
只有符合以下所有标准,参与者才有资格包括在研究中:
[0206]
签署知情同意书时,年龄为40岁或以上;
[0207]
就诊1时出现指数膝疼痛》12周;
[0208]
x光支持根据american college of rheumatology的骨关节炎诊断,具有kellgren-lawrence 2至4射线照相分类的指数膝;具有稳定的血糖控制,如筛查时糖化血红蛋白(hba1c)小于或等于10所示的;
[0209]
是遵守所提供的生育和避孕要求的男性或女性;愿意为研究中的情况停止所有疼痛药物,但在随访期内直至v801允许的抢救药物除外;
[0210]
必须在两个手臂上都有静脉通道用于iv输液和样本采集。
[0211]
疼痛特征:
[0212]
在就诊1和2时,视觉模拟评分(vas)疼痛值》40和《95;
[0213]
基于患者报告或病史每日疼痛史持续至少12周;
[0214]
疼痛灾难性评分值为≤30分;
[0215]
体重:体重指数《40kg/m2(含)
[0216]
知情同意书:
[0217]
能够提供知情同意书,包括遵守知情同意书(icf)和协议中列出的要求和限制;
[0218]
在研究期间,他们是可靠的、愿意的,且能够参与所有必需的协议程序;
[0219]
愿意维持任何正在进行的非药物止痛疗法(例如物理疗法)的一致方案,并且在研究参与期间不会开始任何新的非药物镇痛疗法;
[0220]
在研究期间,愿意为研究中的病症停止所有疼痛药物,除了方案允许的抢救药物;
[0221]
在随机化前的7天中的至少5天,在pdep期间必须输入所需的每日评估。
[0222]
排除标准:
[0223]
如果以下任何标准适用,参与者将被排除在研究之外:
[0224]
基本上或完全失能,且无法完全参与所有协议程序,例如,卧床不起或被限制在轮
椅上,很少或不允许自理;
[0225]
指数膝中存在手术硬件或其他异外来物;
[0226]
具有不稳定的指数关节(如前交叉韧带撕裂);
[0227]
在开始清除期前3个月内对受影响的膝进行了手术或治疗性注射;
[0228]
患有纤维肌痛、慢性疼痛综合征或其他并发的医疗或关节炎疾病,这些疾病可能会影响指数膝关节的评估;
[0229]
具有reiter综合征、类风湿性关节炎、银屑病关节炎、强直性脊柱炎、与炎性肠病相关的关节炎、结节病或淀粉样变的病史;
[0230]
具有指数膝的活动性膝关节感染或晶体疾病的临床症状和体征
[0231]
具有指数关节感染史;
[0232]
具有由于晶体导致的关节炎病史(例如痛风、假性痛风);
[0233]
患有同侧髋关节炎;或
[0234]
其他医疗状况:他们在就诊2后的24周内进行了透明质酸关节内注射;在就诊1或就诊2时,基于ckd-epi公式,egfr小于70ml/min/1.73m2;在就诊3的3个月内,出现任何临床严重或不稳定的心血管、肌肉骨骼疾病、胃肠道、内分泌、血液学、肝脏、代谢、泌尿、肺部、皮肤病、免疫或眼科疾病。
[0235]
既往/伴随治疗:他们接受过任何针对神经生长因子(ngf)的抗体,或针对egfr或egfr酪氨酸激酶抑制剂的抗体;对单克隆抗体有过敏反应史,或具有临床意义的多重或严重药物过敏史,包括但不限于多形性大红斑、线性免疫球蛋白a皮肤病、毒性表皮坏死溶解或剥脱性皮炎;有不受控制的哮喘、湿疹、严重的过敏反应、严重的遗传性血管性水肿或常见的可变免疫缺陷的病史或存在,和
[0236]
生殖:她们是怀孕或哺乳期的妇女。要求参与者在双盲研究期间保持类似的活动水平。不允许开始新的锻炼计划或新的剧烈活动。
[0237]
接受指数膝oa的物理治疗的参与者应保持相同的治疗方案(强度和频率)。
[0238]
研究评估和程序:
[0239]
药代动力学:
[0240]
将采集大约4ml的静脉血样,以测量抗体i浓度。样品将用于评估抗体i的pk。现场人员将记录抗体i施用的日期和时间(24小时时钟时间)(输注开始和结束),以及每个pk样品的日期和时间(24小时时钟时间)。
[0241]
药效学:
[0242]
将采集每种大约4ml的静脉血样,用于测量表皮调节素。任何剩余的血样可用于测试其他潜在pd终点,包括但不限于tgf-α。
[0243]
免疫原性评估:
[0244]
通过经验证的测定评估免疫原性,该测定设计用于在赞助商批准的实验室中检测抗体i存在下的ada。可进一步表征抗体中和抗体i活性的能力。在指定的就诊和时间,将采集给药前静脉血样,以确定针对抗体i的抗体产生。将记录每次样本采集的实际日期和时间(24小时时钟时间)。如果最后一次计划评估或停药就诊时的免疫原性样品为治疗突发的(te)抗药物抗体(ada)阳性,则可采集其他的样品直至信号恢复到基线(即,不再是te-ada阳性)或在最后一次给药后直至1年。为了帮助解释这些结果,将在相同的时间点采集用于
pk分析的给药前血样。
[0245]
统计假设:
[0246]
定义了贝叶斯临界成功因子(csf)并用于评估抗体i是否满足其主要终点。将使用本文所述和本领域技术人员已知的方法,评估csf的主要疗效终点,如通过nrs测量的平均疼痛强度,并在每个研究的双盲部分结束时进行计算。csf的一般形式为:概率(治疗效果《目标效果)》概率阈值。治疗效果将定义为终点时从基线的变化的抗体i估计值-安慰剂估计值。目标效果通常通过文献搜索或临床判断来发现。概率阈值通常被设置为具有期望的治疗效果置信水平,或在一系列可信的、假设的真实药物效应场景下具有期望的操作特性,包括无效效应。其他假设将包括针对本文定义的预定目标和终点的主动干预与安慰剂的比较。研究可以在一个方案中进行,其中考虑了多个研究,并且可以按需共享安慰剂数据。
[0247]
主要假设的判定标准定义为在至少70%置信下抗体i在nrs测量的平均疼痛强度上比安慰剂好至少0.55个单位。关键次要无效假设是抗体i和安慰剂在关键次要终点(疼痛亚尺度评分从基线到终点的平均变化)上没有差异。关键次要假设的判定标准定义为在至少70%置信下抗体i在疼痛子量表上至少比安慰剂好0.35个单位。
[0248]
样本量测定:
[0249]
约125名参与者将分别以2:1的比例随机分配给抗体i和安慰剂。预计约107名参与者将完成本研究的双盲治疗期。该样本大小将提供大于80%的能力,来证明在疼痛子量表(womac=western ontario and mcmaster university骨关节炎指数)上主动治疗臂具有优于安慰剂至少0.35个单位的≥0.7的后验概率。
[0250]
能力计算的假设是,根据womac疼痛子量表测量的,安慰剂和抗体i的疼痛强度从基线到终点平均降低分别约为3个单位和4个单位,共同标准偏差为2.25。如果安慰剂和抗体1之间不存在治疗差异,通过上述有效性标准(即假阳性)的概率约为0.1。能力计算和样本量确定的模拟在facts6.0版中进行。
[0251]
分析人群:
[0252]
药代动力学群包括例如在就诊3时接受全剂量抗体i的所有随机参与者并在就诊4时给药前或就诊4后收集有至少1份可评估pk样品。
[0253]
实施例8:评估抗体i治疗糖尿病性周围神经疼痛的随机的、安慰剂对照的2期临床试验。
[0254]
本研究的目的是提供抗体i缓解糖尿病性周围神经疼痛(dpnp)功效的人类临床证据。将收集数据以评估该研究群中抗体i的安全性和耐受性。还将探讨药代动力学(pk)特性、药效学(pd)效应和免疫原性概况。该概念验证研究的全部数据将评估与抗体i相关的益处和风险,并为抗体i的临床开发提供信息。
[0255]
总体设计:
[0256]
这是一项为期26周、2期、随机、双盲、安慰剂对照研究,在dpnp参与者中对抗体i与安慰剂进行了比较。这是一项随机、研究者和参与者盲、安慰剂对照的2期临床试验。大约125名参与者将被随机分配到研究干预组(84名抗体i和41名安慰剂)。这项为期26周的研究包括8周的双盲治疗期和18周的随访期。该研究设计如图2所示。
[0257]
研究干预将通过不知情人员在约1小时内缓慢静脉(iv)输注进行。如果观察到输
注反应,则可根据需要降低输注速率。剂量为750mg起始剂量,然后每2周iv 500mg,共4剂。给药制剂是用无菌水0.9%氯化钠溶液配制的冻干粉。参与者将每2周接受一次iv输注,共输4次。每次输注完成后,将对参与者进行至少4小时的监测。
[0258]
在患者就诊时,完成所有治疗后样本收集和安全性监测,并指示参与者在就诊解除前继续完成研究限制和数字评分量表(nrs)日记条目。基于抗体i的长pk半衰期和潜在的持续靶向结合,将在最后一次给药后直至6周内收集疗效数据。将在最后一次干预给药后直至20周内收集安全性、药代动力学(pk)、药效学(pd)和免疫原性样品,以表征安全性和临床免疫原性概况。如果参与者已完成研究的所有要求阶段,包括最后一个预定程序,则认为其已完成本研究。
[0259]
目标和终点:
[0260]
主要和次要目标和终点如下所述。
[0261][0262][0263]
视觉模拟量表(vas):疼痛的vas将在筛查和每次临床就诊时使用。这是一个图形化的单项目量表,其中参与者被要求描述过去一周在研究区域的疼痛强度,范围为0到100:0=无疼痛,100=最严重的可想象的疼痛。参与者通过在描述其疼痛强度的点处放置一条垂直于vas线的线来完成vas。
[0264]
数值评定量表(nrs):在初步数据输入阶段(pdep)期间和整个研究期间每天使用nrs来描述疼痛严重程度。这是一个数字单项目量表,其中参与者被要求描述过去24小时内他们的平均和最严重的疼痛,范围为0到10:0=没有疼痛,10=可以想象的最严重的疼痛程度。参与者每天使用带回家的设备完成nrs。每次临床就诊时都会对参与者的依从性进行审查。将每天收集nrs最严重的疼痛。其他次要测量的得分将在按照就诊时间表规定的每次就诊时进行收集。主要结果测量是根据nrs项目“通过选择描述过去24小时内[研究区域]平均
疼痛水平的数字[0-10]对疼痛进行评分”评估从基线到终点平均疼痛强度的平均变化。选择这项测量是基于其检测疼痛变化的能力及其在不同疾病状态下的常用性。第二个测量是通过nrs项目“请通过选择一个数字[0-10]来对疼痛评级,该数字描述了过去24小时内[研究区域]疼痛的最严重程度”衡量的最严重疼痛强度从基线到终点的平均变化。每天收集每位参与者过去24小时内平均疼痛和最严重疼痛的nrs值。对于统计分析,将针对每周间隔和每两周间隔计算过去24小时内平均和最严重疼痛的平均nrs值。如果参与者完成研究的安慰剂对照部分,则每周间隔的nrs平均值将导致每名参与者8次基线后观察,并且每两周间隔的平均将导致每名参与者4次基线后观测。除非分析计划中另有规定,否则nrs每周间隔的平均值将用于主要疗效分析和下文所述的其他分析。参与者必须在预先指定的时间间隔内拥有每日nrs值的50%或更高,才能计算平均nrs值;否则,该就诊的平均nrs值将被视为缺失。
[0265]
参与者对总体改善的评分:将患者总体变化印象(pgi)用于所有疾病状态。除了整体改善的子方面之外,它还捕捉了参与者的治疗观点。这是一个从1到7的数字量度:1=非常好,而7=非常差。参与者被要求“与你开始服用这种药物之前的情况相比,在最能描述你现在疼痛症状的方框上做标记。”[0266]
情绪功能评估:eq-5d-5l健康状况问卷用于所有疾病状态。eq-5d-5l是解决生活质量问题的最受欢迎的患者完成的工具之一(buchholz i,janssen mf,kohlmann t,feng ys。a systematic review of studies comparing the measurement properties of the three-level and five-level versions of the eq-5d.pharmacoeconomics.2018;36(6):645-661)。它是一个描述系统,其包括5个维度:行动、自我照顾、日常活动、疼痛/不适和焦虑/抑郁。参与者被要求“勾选一个最能描述你今天健康状况的选项”,从每个维度下提供的5个选项中进行选择。5个维度上的得分可以作为健康概况显示或转换为单个汇总索引编号。eq-5d-5l还包括eq vas,它在0到100的垂直vas上记录参与者的自评健康状况:0=你能想象到的最差健康状况,100=你能想到的最佳健康状况。eq-5d-5l版本中使用的工具是一个简短、可靠、经过验证、易于完成的量表,具有卓越的重新测试可靠性,以解决与一些疾病引起的疼痛相关的生活质量。
[0267]
睡眠质量:睡眠障碍是疼痛研究中的一个重要问题。在各种可用的工具中,医学结果研究(mos)睡眠量表为睡眠障碍提供了一个独特的、经心理学验证的分数。这一量表包含了过去一周的12个问题。参与者报告分为6个等级的每种睡眠症状或问题出现的频率,等级从“所有时间”到“没有时间”。关于入睡时间和睡眠量的问题被报告为每晚平均睡眠小时数。该量表管理负担低,已用于不同的疼痛研究,并已在神经性疼痛患者中得到验证。
[0268]
安全性评估:根据典型实践确定安全性评估的计划时间点,并且包括体检、生命体征和体重测量、12导联ecg、临床实验室测试、肝脏安全性监测、c-ssrs和自发报告的aes。
[0269]
功效评估:
[0270]
[0271][0272]
在表征慢性疼痛时,使用一组核心评估和领域,其是:疼痛、身体机能、情绪功能、参与者总体改善评分、不良事件(aes)和参与者处置。选择nrs作为主要终点是基于其已证明的检测疼痛变化的能力及其在所研究的疾病状态中的常用性。
[0273]
简易疼痛量表-修订简表(bpi-sf)
[0274]
bpi-sf是一种数字评分量表,其评估不同疾病状态下疼痛的严重性(严重性量表)、对日常功能的影响(干扰量表)以及疼痛的其他方面(例如,疼痛的位置、药物缓解)(cleeland cs,ryan km.pain assessment:global use of the brief pain inventory.ann acad med singapore.1994年3月;23(2):129-138)。该表描述了bpi修订版中使用的疼痛量表和相应的数字评分量表,该表针对糖尿病多发性神经病的疼痛进行了验证。参与者将评估他们的疼痛严重性,以及在过去24小时内,疼痛如何干扰本表中所述的活动。
[0275]
[0276][0277]
活动时间表(soa):
[0278]
该活动时间表显示了抗体i研究的某些就诊和程序。“主方案”是指在给定疾病状态或多种疾病状态下,为指导数个潜在研究而制定的方案,疾病状态附录是指特定疾病状态的指南,而干预特定附录是指方案中与研究中的给定干预特异性地相关的部分。
[0279]
[0280][0281][0282]
缩写:dsa=疾病状态附录;ed=提前终止;v=就诊.
[0283]a如果减少了参与者的负担,则可在随机化前的其他时间点进行筛选评估。
[0284]b站点确定参与者当前服用的每种疼痛药物的半衰期,以便安排就诊2。由于需要7天的pdep,就诊2可安排在不早于就诊3随机化前的7天
[0285]c疼痛药物的5个半衰期清除期必须在pdep之前,因此大多数参与者至少需要10天。
[0286]
研究群:
[0287]
如果基于患者报告或病史有每日疼痛史,则男性和女性参与者有资格纳入研究。估计的gfr将用于分层,以确保肾影响分析的参与者数量平衡。将在就诊2时收集测量值,且子集大于或等于90,且小于90。
[0288]
密歇根神经病变筛查工具用于评估糖尿病患者腿部和足部的神经病变(the michigan neuropathy screening instrument.密西根大学网站,可访问:http://www.med.umich.edu/borc/profs/documents/svi/mnsi_patient.pdf.published 2000.http://www.med.umich.edu/borc/profs/documents/svi/mnsi_patient.pdf.2000年公开。2019年12月11日访问)。将根据活动时间表进行管理。本表描述了该工具中包含的评估。
[0289]
部分评估由
……
完成项目数a病史研究参与者15
b身体保健专业人员5
[0290]
将同时进行a部分和b部分。只有b部分将用于确定纳入研究。
[0291]
纳入标准:
[0292]
只有符合以下所有标准,参与者才有资格参与研究:
[0293]
签署知情同意书时为18岁或更大年龄;
[0294]
每天都有继发于周围神经病变的对称性足痛,存在至少6个月,并如通过使用密歇根神经病变筛查工具b部分诊断为≥3(密歇根大学[www]);
[0295]
具有1型或2型糖尿病的病史和当前诊断;
[0296]
具有稳定的血糖控制,如筛选时通过糖化血红蛋白≤11所示的;
[0297]
遵守所提供的生育和避孕要求的男性或女性;
[0298]
他们愿意为所研究的病症停止所有疼痛药物,除了随访期内直至v801允许的抢救药物;
[0299]
必须在两个手臂上都有静脉通道用于静脉输液和样品采集。
[0300]
疼痛特征:
[0301]
在就诊1和就诊2时,视觉模拟评分(vas)疼痛值》40和《95;
[0302]
基于患者报告或病史,每日疼痛史持续至少12周;
[0303]
疼痛灾难性评分具有≤30的值;
[0304]
体重:体重指数《40kg/m2(含)。
[0305]
知情同意书:
[0306]
能够提供知情同意书,包括遵守知情同意书(icf)和协议中列出的要求和限制;
[0307]
在研究期间,他们是可靠的、愿意的,且能够参与所有必需的协议程序;
[0308]
愿意维持任何正在进行的非药物止痛疗法(例如物理疗法)的一致方案,并且在研究参与期间不会开始任何新的非药物镇痛疗法;
[0309]
在研究期间,愿意为研究中的病症停止所有疼痛药物,除了方案允许的抢救药物;
[0310]
在随机化前的7天中的至少5天,在pdep期间必须输入所需的每日评估。
[0311]
排除标准:
[0312]
如果以下任何标准适用,参与者将被排除在研究外:
[0313]
例如,由于某些类型的化疗,或其他类型的周围神经病变,他们患有目前的药物诱发的神经病变;
[0314]
患有已知的遗传性运动、感觉或自主神经病变。
[0315]
其他医疗条件:
[0316]
在就诊1或就诊2时,基于ckd-epi公式,其egfr小于70ml/min/1.73m2;
[0317]
在就诊3的3个月内,出现任何临床严重或不稳定的心血管、肌肉骨骼疾病、胃肠道、内分泌、血液学、肝脏、代谢、泌尿、肺部、皮肤病、免疫或眼科疾病。
[0318]
既往/伴随治疗:接受过任何针对神经生长因子(ngf)的抗体,或针对egfr或egfr酪氨酸激酶抑制剂的抗体;对单克隆抗体有过敏反应史,或具有临床意义的多重或严重药物过敏史,包括但不限于多形性红斑、线性免疫球蛋白a皮肤病、毒性表皮坏死溶解或剥脱性皮炎;有不受控制的哮喘、湿疹、严重的过敏反应、严重的遗传性血管性水肿或常见的可变免疫缺陷的病史或存在上述疾病,和
[0319]
生殖:她们是怀孕或哺乳期的妇女。
[0320]
要求参与者在双盲研究期间保持类似的活动水平。不允许开始新的锻炼计划或新的剧烈活动。
[0321]
药代动力学:
[0322]
将采集大约4ml的静脉血样,用于测量抗体i浓度。样品将用于评估抗体i的pk。现场人员将记录抗体i给药的日期和时间(24小时时钟时间)(输注开始和结束),以及每个pk样品的日期和时间(24小时时钟时间)。
[0323]
药效学:
[0324]
将采集每种大约4ml的静脉血样,用于测量表皮调节素。任何剩余的血样可用于测试其他潜在pd终点,包括但不限于tgf-α。
[0325]
免疫原性评估:
[0326]
将通过经验证的测定评估免疫原性,该测定设计用于在赞助商批准的实验室中检测抗体i存在下的ada。可进一步表征抗体中和抗体i活性的能力。在指定的就诊和时间,将采集给药前静脉血样,以确定针对抗体i的抗体产生。将记录每次样本采集的实际日期和时间(24小时时钟时间)。如果最后一次计划评估或停药就诊时的免疫原性样品为治疗突发的(te)抗药物抗体(ada)阳性,则可采集其他的样品直至信号恢复到基线(即,不再是te-ada阳性)或在最后一次给药后直至1年。为了帮助解释这些结果,将在相同的时间点采集用于pk分析的给药前血样。
[0327]
统计假设:
[0328]
统计假设:
[0329]
定义了贝叶斯临界成功因子(csf)并用于评估抗体i是否满足其主要终点。将使用本文所述和本领域技术人员已知的方法,评估csf的主要疗效终点,如通过nrs测量的平均疼痛强度,并在每个研究的双盲部分结束时进行计算。csf的一般形式为:概率(治疗效果《目标效果)》概率阈值。治疗效果将定义为终点时从基线的变化的抗体i估计值-安慰剂估计值。目标效果通常通过文献搜索或临床判断来发现。概率阈值通常被设置为治疗效果具有期望的置信水平,或在一系列可信的、假设的真实药物效应场景下具有期望的操作特性,包括无效效应。其他假设将包括针对本文定义的预定目标和终点的主动干预与安慰剂的比较。研究可以在一个方案中进行,其中考虑了多个研究,并且可以按需共享安慰剂数据。
[0330]
主要假设的判定标准定义为至少70%的置信度,即抗体i在nrs测量的平均疼痛强度上比安慰剂好至少0.4个单位。
[0331]
bpi-sf持续疗效分析:将进行贝叶斯纵向混合效应模型重复测量(mmrm)分析,以评估总疼痛干扰量表从基线到每次基线后就诊的变化。该分析将用于分析从基线到每次基线后就诊的变化:个体疼痛干扰、总疼痛干扰(7种反应的总和)和个体疼痛严重程度评分。将在每个基线后时间点估计每个治疗组中达到预定二元疗效结果的参与者比例,并用于与治疗组比较。将通过拟合包括所有基线后观察的贝叶斯纵向模型来提供估计值。预先指定的二元疗效结果包括通过bpi-sf个体严重性评分测量的从基线降低》30%、50%和70%的参与者比例,以及通过bpi-sf-总干扰评分测量的从基线降低》30%、50%、70%的参与者比例。
[0332]
样本量测定:
[0333]
约125名参与者将分别以2:1的比例随机分配给抗体i和安慰剂。预计大约107名参与者将完成本研究的双盲治疗期。该样本大小将提供大于80%的能力,以证明主动治疗臂具有≥0.7后验概率,如通过nrs测量的,平均疼痛强度比安慰剂好至少0.4个单位。
[0334]
能力计算的假设是,如通过nrs测量的,安慰剂和抗体i的平均疼痛强度在终点分别约为1.58单位和2.58单位,共同标准差为2。如果安慰剂和抗体i之间不存在治疗差异,通过上述有效性标准(即假阳性)的概率约为0.06。能力计算和样本量确定的模拟在facts 6.0版中进行。
[0335]
分析群:
[0336]
药代动力学群包括例如在就诊3时接受全剂量抗体i的所有随机参与者并在就诊4给药前或就诊4后收集至少1份可评估pk样品。
[0337]
实施例9:评估抗体i治疗慢性腰痛的随机安慰剂对照2期临床试验。
[0338]
本研究的目的是为抗体i缓解慢性腰痛(clbp)引起的疼痛提供人类临床证据。将收集数据以评估该研究群中抗体i的安全性和耐受性。还将探讨药代动力学(pk)特性、药效学(pd)效应和免疫原性概况。该概念验证研究的全部数据将评估与抗体i相关的益处和风险,并为抗体i的临床开发提供信息。
[0339]
总体设计:
[0340]
这是一项26周、2期、随机、双盲、安慰剂对照的研究,将在患有clbp的参与者中比较抗体i vs安慰剂。这是一项随机、研究者和参与者盲、安慰剂对照的2期临床试验。大约150名参与者将被随机分配到研究干预组(100名在抗体i组和50名在安慰剂组)。这项为期26周的研究包括8周的双盲治疗期和18周的随访期。该研究设计如图2所示。注意,方法、规范和程序可能与实施例7、8和9相同,且本领域技术人员将认识到,无需在每个实施例中重复这些,并且类似地,理解针对健康和不良事件的额外常规测试,因为是临床研究典型的,是实施例7、8和9的研究的设计和执行的一部分。
[0341]
研究干预将通过不知情人员在约1小时内缓慢静脉(iv)输注进行。如果观察到输注反应,则可根据需要降低输注速率。剂量为750mg起始剂量,然后每2周静脉注射500mg,共4剂。给药制剂是用无菌水0.9%氯化钠溶液配制的冻干粉。参与者将每2周接受一次静脉输液,共输4次。每次输注完成后,将对参与者进行至少4小时的监测。
[0342]
在患者就诊时,完成所有治疗后样本收集和安全性监测,并指示参与者在就诊解除前继续完成研究限制和数字评分量表(nrs)日记条目。根据抗体i的长pk半衰期和潜在的持续靶向结合,将在最后一次给药后直至6周内收集疗效数据。将在最后一次干预给药后直至20周收集安全性、药代动力学(pk)、药效学(pd)和免疫原性样本,以表征安全性和临床免疫原性概况。如果参与者已完成研究的所有要求阶段,包括最后一个计划程序,则认为其已完成本研究。
[0343]
目标和终点:
[0344]
疗效结果测量:主要结果测量包括:数值评分量表(nrs)测量的平均疼痛强度从基线的变化,nrs测量的平均疼痛强度从基线的变化[时间范围:基线,至第8周],次要结果测量包括:roland morris失能问卷(rmdq)从基线的变化,rmdq从基线的变化[时间范围:基线,至第8周],患者总体变化印象测量的从基线总体改善的变化,患者总体变化印象测量的总体改善从基线的变化[时间范围:基线,至第8周],nrs测量的最剧烈疼痛强度从基线的变
化,nrs测量的最剧烈疼痛强度从基线的变化[时间范围:基线,至第8周],疼痛视觉模拟量表(vas)从基线的变化,疼痛vas从基线的变化[时间范围:基线,至第八周],医疗结果研究睡眠量表(mos睡眠量表)从基线的变化,mos睡眠量表中睡眠量表从基线的变化[时间范围:基线,至第8周],救援药物总量救援药物总量[时间范围,基质至第八周],euroqol-5d 5级问卷(eq-5d-5l)从基线的变化,eq-5d-5l从基线的变化。
[0345]
评估概述
[0346][0347]
功效评估:
[0348]
[0349][0350]
在表征慢性疼痛时,使用一组核心评估和领域,其是:疼痛、身体机能、情绪功能、参与者总体改善评分、不良事件(aes)和参与者处置。选择nrs作为主要终点是基于其已证明的检测疼痛变化的能力及其在研究疾病状态中的常用性。
[0351]
视觉模拟量表(vas):疼痛的vas将在筛查和每次临床就诊时使用。这是一个图形化的单项目量表,其中参与者被要求描述过去一周在研究区域的疼痛强度,范围为0到100:0=无疼痛,100=最严重的可想象的疼痛。参与者通过在描述其疼痛强度的点处放置一条垂直于vas线的线来完成vas。
[0352]
数值评定量表(nrs):在初步数据输入阶段(pdep)期间和整个研究期间每天使用nrs来描述疼痛严重程度。这是一个数字单项目量表,其中参与者被要求描述过去24小时内他们的平均和最严重的疼痛,范围为0到10:0=没有疼痛,10=可以想象的最严重的疼痛程度。参与者每天使用带回家的设备完成nrs。每次临床就诊时都会对参与者的依从性进行审查。将每天收集nrs最严重的疼痛。其他次要测量的得分将在按照就诊时间表规定的每次就诊时进行收集。主要结果测量是根据nrs项目“通过选择描述过去24小时内[研究区域]平均疼痛水平的数字[0-10]对疼痛进行评分”评估从基线到终点平均疼痛强度的平均变化。选择这项测量是基于其检测疼痛变化的能力及其在不同疾病状态下的常用性。第二个测量是通过nrs项目“请通过选择一个数字[0-10]来对疼痛评级,该数字描述了过去24小时内[研究区域]疼痛的最严重程度”衡量的最严重疼痛强度从基线到终点的平均变化。每天收集每位参与者过去24小时内平均疼痛和最严重疼痛的nrs值。对于统计分析,将针对每周间隔和
每两周间隔计算过去24小时内平均和最严重疼痛的平均nrs值。如果参与者完成研究的安慰剂对照部分,则每周间隔的nrs平均值将导致每名参与者8次基线后观察,每两周间隔的平均将导致每名参与者4次基线后观测。除非分析计划中另有规定,否则nrs每周间隔的平均值将用于主要疗效分析和下文所述的其他分析。参与者必须在预先指定的时间间隔内拥有每日nrs值的50%或更高,才能计算平均nrs值;否则,该就诊的平均nrs值将被视为缺失。
[0353]
参与者对总体改善的评分:将患者总体变化印象(pgi)用于所有疾病状态。除了整体改善的子方面之外,它还捕捉了参与者的治疗观点。这是一个从1到7的数字量度:1=非常好,而7=非常差。参与者被要求“与你开始服用这种药物之前的情况相比,在最能描述你现在疼痛症状的方框上做标记。”[0354]
情绪功能评估:eq-5d-5l健康状况问卷用于所有疾病状态。eq-5d-5l是解决生活质量问题的最受欢迎的患者完成的工具之一(buchholz i,janssen mf,kohlmann t,feng ys。a systematic review of studies comparing the measurement properties of the three-level and five-level versions of the eq-5d.pharmacoeconomics.2018;36(6):645-661.)。它是一个描述系统,其包括5个维度:行动、自我照顾、日常活动、疼痛/不适和焦虑/抑郁。参与者被要求“勾选一个最能描述你今天健康状况的选项”,从每个维度下提供的5个选项中进行选择。5个维度上的得分可以作为健康概况显示或转换为单个汇总索引编号。eq-5d-5l还包括eq vas,它在0到100的垂直vas上记录参与者的自评健康状况:0=你能想象到的最差健康状况,100=你能想到的最佳健康状况。eq-5d-5l版本中使用的工具是一个简短、可靠、经过验证、易于完成的量表,具有卓越的重新测试可靠性,以解决与一些疾病引起的疼痛相关的生活质量。
[0355]
睡眠质量:睡眠障碍是疼痛研究中的一个重要问题。在各种可用的工具中,医学结果研究(mos)睡眠量表为睡眠障碍提供了一个独特的、经心理学验证的分数。这一量表包含了过去一周的12个问题。参与者报告分为6个等级的每种睡眠症状或问题出现的频率,等级从“所有时间”到“没有时间”。关于入睡时间和睡眠量的问题被报告为每晚平均睡眠小时数。
[0356]
安全性评估:根据典型实践确定安全性评估的计划时间点,并且包括体检、生命体征和体重测量、12导联ecg、临床实验室测试、肝脏安全性监测、c-ssrs和自发报告的aes。
[0357]
roland morris失能问卷:roland morriss失能问卷(rmdq)是一种简单、灵敏、可靠的方法,用于测量背痛患者的失能。rmdq由24项与患者对背痛和相关失能的认知有关的陈述,这些陈述基于:身体能力/活动、睡眠/休息、心理社会、家庭管理、饮食和疼痛频率。参与者被问到他们是否觉得该陈述描述了他们当天的情况。通过计算“是”回答的数量得出总分,范围从:0=无失能至24=最大失能。
[0358]
paindetect:paindetect问卷由7个关于神经性疼痛症状质量的问题组成。最初提出在腰痛患者中捕捉神经性疼痛表型(freynhagen 2006)。参与者很容易回答,并且不需要体检。得分为≥19表明疼痛可能表现为神经性(》90%)。
[0359]
统计分析:
[0360]
该表定义了慢性腰痛的分层因素。
[0361]
神经痛paindetect评分肯定存在≥19不清楚或不存在《19
[0362]
isa中可定义任何其他分层因素。
[0363]
终点分析-rmdq
[0364]
持续功效分析:将进行贝叶斯纵向混合效应模型重复测量(mmrm)分析,以评估rmdq评分从基线到每次基线后就诊的变化。将按照主方案中的描述实施相同的安慰剂借用策略;然而,不会从其他dsa借用治疗效果。还将仅使用来自各自isa的数据进行额外的贝叶斯mmrm分析,isa不使用任何借用的安慰剂信息。该分析将用于分析rmdq评分从基线到每次基线后就诊的变化。(参见freynhagen r,baron r,gockel u,tr.paindetect:a new screening questionnaire to identify neuropathic components in patients with back pain.curr med res opin.curr med res opin.2006;22(10):1911-1920.)
[0365]
此表描述了模型中包含的信息。
[0366][0367]
分类功效分析:
[0368]
每个治疗组中达到预定二元疗效结果的参与者比例将在每个基线后时间点进行估计,并用于与治疗组进行比较。估计值将通过拟合贝叶斯纵向模型提供,该模型包括所有基线后观测。
[0369]
预先指定的二元疗效结果包括isa中参与者的比例:
[0370]
·
如通过rmdq评分测量的从基线降低大于30%、50%和70%,和
[0371]
·
rmdq评分从基线降低至少3.5个点。
[0372]
该模型将包括上述连续疗效分析模型中描述的分类和连续协变量,但不使用基线和就诊的交互作用。将为每个治疗组提供rmdq评分从基线到终点变化百分比的累积分布函数。
[0373]
活动时间表(soa):
[0374]
本活动计划表显示了抗体i研究的某些就诊和程序。“主方案”是指在给定疾病状态或多种疾病状态下指导若干潜在研究的方案设置,并且干预特定附录是指与研究中的特定干预相关的方案部分。
[0375]
[0376][0377]
[0378][0379]
缩写:dsa=疾病状态附录;ed=提前终止;v=就诊
[0380]a如果减少了参与者的负担,则可在随机化前的其他时间点进行筛选评估。
[0381]b站点确定参与者当前服用的每种疼痛药物的半衰期,以便安排第2次就诊。由于需要7天pdep,就诊2安排在不早于就诊3随机化前7天。
[0382]
c疼痛药物的5个半衰期清除期必须在pdep之前,因此大多数参与者至少需要10天。
[0383]
研究人群:
[0384]
如果基于患者报告或病史有每日疼痛史,则男性和女性参与者有资格纳入研究。
[0385]
知情同意书:
[0386]
能够提供知情同意书,包括遵守知情同意书(icf)和协议中列出的要求和限制;在研究期间,他们是可靠的、愿意的,且能够参与所有必需的协议程序;愿意维持任何正在进行的非药物止痛疗法(例如物理疗法)的一致方案,并且在研究参与期间不会开始任何新的非药物镇痛疗法;在研究期间,愿意为研究中的病症停止所有疼痛药物,除了方案允许的抢救药物;在随机化前的7天中的至少5天,在pdep期间必须输入所需的每日评估。
[0387]
纳入标准:
[0388]
只有符合以下所有标准,参与者才有资格包括在研究中:
[0389]
·
签署知情同意书时,年龄为18岁或以上;
[0390]
·
在筛选时,视觉模拟评分(vas)疼痛值≥40和《95;
[0391]
·
基于患者报告或病史每日疼痛史持续至少12周;
[0392]
·
疼痛灾难性评分值为≤30分;
[0393]
·
体重指数《40kg/m2(含)
[0394]
·
愿意维持任何正在进行的非药物止痛疗法(例如物理疗法)的一致方案,并且在研究参与期间不会开始任何新的非药物镇痛疗法;
[0395]
·
在研究期间,愿意为研究中的病症停止所有疼痛药物,除了方案允许的抢救药物;
[0396]
·
有至少3个月的腰痛病史,位于第12胸椎和臀下褶皱之间,使用或未使用辐射。
[0397]
·
通过quebec task force category分类的1至3级腰痛病史。
[0398]
·
血糖控制稳定,如通过筛查时糖化血红蛋白(hba1c)低于或等于10所示的。
[0399]
·
筛查期间,估计肾小球滤过率(egfr)小于70ml/min/1.73m2。
[0400]
·
男性或女性能够遵守生殖和避孕要求。
[0401]
排除标准:
[0402]
如果以下任何标准适用,参与者将被排除在研究之外:
[0403]
·
基线后3个月内出现任何临床严重或不稳定的心血管、肌肉骨骼疾病、胃肠道
(gi)、内分泌、血液学、肝脏、代谢、泌尿、肺部、皮肤病、免疫或眼科疾病。
[0404]
·
接受了任何针对神经生长因子(ngf)的抗体、针对egfr的抗体或egfr酪氨酸激酶抑制剂(tki)。
[0405]
·
有单克隆抗体过敏反应史,或有临床意义的多重或严重药物过敏史,包括但不限于多形性红斑、线性免疫球蛋白a皮肤病、毒性表皮坏死溶解或剥脱性皮炎。
[0406]
·
有不受控制的哮喘、湿疹、严重过敏、严重遗传性血管性水肿或常见的可变免疫缺陷病史或存在。
[0407]
·
在开始清除期前的3个月内使用过治疗性注射(肉毒杆菌毒素或皮质类固醇);
[0408]
·
有骨质疏松性压缩性骨折的病史或目前有骨质疏松性压缩性骨折;最近有过重大创伤(v3后6个月内);
[0409]
·
在过去6个月内曾接受过手术干预以治疗腰痛。
[0410]
·
生殖:怀孕或哺乳期妇女。
[0411]
·
既往/伴随治疗:他们接受过任何针对神经生长因子(ngf)的抗体,或针对egfr或egfr酪氨酸激酶抑制剂的抗体;对单克隆抗体有过敏反应史,或具有临床意义的多重或严重药物过敏史,包括但不限于多形性红斑、线性免疫球蛋白a皮肤病、毒性表皮坏死溶解或剥脱性皮炎;有未控制的哮喘、湿疹、严重的过敏反应、严重的遗传性血管性水肿或常见的可变免疫缺陷病史或存在未控制的哮喘、湿疹、严重的过敏反应、严重的遗传性血管性水肿或常见的可变免疫缺陷病史,和
[0412]
研究评估和程序:
[0413]
药代动力学:
[0414]
将采集约4ml的静脉血液样本,用于测量抗体i浓度。样本将用于评估抗体i的pk。现场人员将记录抗体i给药(输注开始和结束)的日期和时间(24小时时钟时间),以及每个pk样本的日期和日期(24小时时钟时间)。
[0415]
药效学:
[0416]
将采集每种约4ml的静脉血液样本,用于测量表皮调节素。任何剩余的血液样本可用于测试其他潜在pd终点,包括但不限于tgf-α。
[0417]
免疫原性评估:
[0418]
将通过经验证的测定评估免疫原性,该测定设计用于在赞助商批准的实验室中检测抗体i存在下的ada。可进一步表征抗体中和抗体i活性的能力。在指定的就诊和时间,将采集给药前静脉血样,以确定针对抗体i的抗体产生。将记录每次样本采集的实际日期和时间(24小时时钟时间)。如果最后一次计划评估或停药就诊时的免疫原性样品为治疗突发的(te)抗药物抗体(ada)阳性,则可采集其他的样品直至信号恢复到基线(即,不再是te-ada阳性)或在最后一次给药后直至1年。为了帮助解释这些结果,将在相同的时间点采集用于pk分析的给药前血样。
[0419]
统计假设:
[0420]
定义了贝叶斯临界成功因子(csf)并用于评估抗体i是否满足其主要终点。将使用本文所述和本领域技术人员已知的方法,评估csf的主要疗效终点,如通过nrs测量的平均疼痛强度,并在每个研究的双盲部分结束时进行计算。csf的一般形式为:概率(治疗效果《目标效果)》概率阈值。治疗效果将定义为终点时从基线的变化的抗体i估计值-安慰剂估计
值。目标效果通常通过文献搜索或临床判断来发现。概率阈值通常被设置为治疗效果具有期望的置信水平,或在一系列可信的、假设的真实药物效应场景下具有期望的操作特性,包括无效效应。其他假设将包括针对本文定义的预定目标和终点的主动干预与安慰剂的比较。研究可以在一个方案中进行,其中考虑了多个研究,并且可以按需共享安慰剂数据。
[0421]
主要假设的判定标准定义为在至少70%置信下抗体i在nrs测量的平均疼痛强度上比安慰剂好至少0.55个单位。关键次要无效假设是抗体i和安慰剂在关键次要终点(相关疼痛评分从基线到终点的平均变化)上没有差异。关键次要假设的判定标准定义为在至少70%置信下抗体i在相关疼痛评分上至少比安慰剂好0.35个单位。
[0422]
用于分析的人群:
[0423]
药代动力学人群包括例如在就诊3接受全剂量抗体i的所有随机参与者,并且在就诊4给药前或就诊4后收集了至少1个可评估的pk样本。
[0424]
列出氨基酸和核酸序列
[0425]
重链cdr
[0426]
seqidno:1gytftdayin
[0427]
seqidno:2wiwpgpvityynpkfkg
[0428]
seqidno:3revlspfay
[0429]
轻链cdr
[0430]
seqidno:4rssqsivhstgntyle
[0431]
seqidno:5kvsnrfs
[0432]
seqidno:6fhgthvpyt
[0433]
重链可变区
[0434]
seqidno:7(抗体i和抗体ii)
[0435][0436]
seqidno:8(抗体iii)
[0437][0438]
轻链可变区
[0439]
seqidno:9(抗体i)
[0440][0441]
seqidno:10(抗体ii)
[0442][0443]
seqidno:11(抗体iii)
[0444][0445]
完整重链
[0446]
seq id no:12(抗体i和抗体ii)
[0447][0448]
完整轻链
[0449]
seq id no:13(抗体i)
[0450][0451]
seq id no:14(抗体ii)
[0452][0453]
核苷酸序列
[0454]
重链可变区
[0455]
seq id no:15
[0456][0457]
核苷酸序列
[0458]
轻链可变区
[0459]
seq id no:16
[0460][0461]
seq id no:17
[0462][0463]
成熟人tgfα
[0464]
seq id no:18
[0465][0466]
成熟小鼠(mus musculus)tgfα
[0467]
seq id no:19
[0468][0469]
成熟大鼠(rattus norvegicus)tgfα
[0470]
seq id no:20
[0471][0472]
成熟食蟹猴(macaca fascicularis)tgfαseq id no:21
[0473][0474]
成熟人表皮调节素
–
添加n-末端甲硫氨酸
[0475]
seq id no:22
[0476][0477]
成熟小鼠(musmusculus)表皮调节素
–
添加n-末端甲硫氨酸
[0478]
seq id no:23
[0479][0480]
成熟食蟹猴(macacafascicularis)表皮调节素
[0481]
seq id no:24
[0482][0483]
成熟人epigen
[0484]
seq id no:25
[0485][0486]
成熟小鼠(mus musculus)epigen
[0487]
seq id no:26
[0488][0489]
成熟人egf
–
添加n-末端甲硫氨酸
[0490]
seq id no:27
[0491][0492]
成熟人hbegf
[0493]
seq id no:28
[0494][0495]
成熟人betacellulin
[0496]
seq id no:29
[0497][0498]
成熟人amphiregulin
[0499]
seq id no:30
[0500][0501]
抗体iii完整重链
–
小鼠抗体
[0502]
seq id no:31
[0503][0504]
抗体iii完整轻链
–
小鼠抗体
[0505]
seq id no:32
[0506][0507]
成熟人表皮调节素
[0508]
seq id no:33
[0509]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1