本发明涉及经口给予制剂,其含有曲马多或其药学上可接受的盐作为有效成分,且副作用出现率低。更详细而言,本发明涉及经口给予制剂,通过使经口给予时的曲马多未变化体和活性代谢物的血药浓度等落入一定的范围内,从而具有优异的有效性、且副作用出现率得以降低。
背景技术:
1、曲马多〔化学名:(1rs,2rs)-2-[(二甲基氨基)甲基]-1-(3-甲氧基苯基)环己醇〕是1962年由德国grünenthal gmbh合成的化合物。作为阿片系镇痛药,目前世界上有一百多个国家以各种剂型(经口剂、栓剂、注射剂、复方制剂等)持续贩卖,因此可知是长年受到市场青睐的极其优异的药剂。需要说明的是,无论剂型如何,曲马多制剂的有效成分通常为曲马多盐酸盐。
2、曲马多是who方式癌症疼痛治疗中被分类为第2阶段药的非麻药性的弱阿片。在日本保险中也适用于非癌症性慢性疼痛,作为通用性高的镇痛剂使用。曲马多本身仅具有较弱的去甲肾上腺素/血清素再摄入抑制作用,但在生物体内被脱甲基化,被代谢为显示出弱的μ-阿片受体活性的活性代谢产物(主要为后述的m1)。通过该较弱的μ-阿片受体活性和去甲肾上腺素再摄入抑制作用的协同作用而发挥良好的镇痛作用,认为对伤害性疼痛、神经障碍性疼痛均示出有效性。
3、曲马多是具有氨基的酚醚类的合成化合物,是当量包含(+)-曲马多和(-)-曲马多的镜像异构体的外消旋体混合物。曲马多为水溶性,通过经口给予而被迅速地几乎完全地吸收,经口用的胶囊剂的情况,在给药1.6~1.9小时后达到血药浓度峰值,半衰期约为5~6小时。另一方面,活性代谢物o-去甲基曲马多(m1)在给药2小时后达到血药浓度峰值,半衰期为6~7小时。经口给予的曲马多在脑内10分钟内达到峰值浓度,但m1代谢物在20~60分钟后达到峰值浓度。另外,在血中曲马多/m1代谢物比为0.5~1.0,m1代谢物浓度高,但是m1代谢物的中枢转移性低于曲马多,因此在脑内曲马多以比m1代谢物高4~15倍的浓度存在。
4、作为曲马多的代谢途径,主要受细胞色素p450(cyp)2d6的o-脱甲基化、另外一部分受cyp3a4的n-脱甲基化反应(第一相反应),这些脱甲基化体被葡糖醛酸结合或硫酸结合(第二相反应)。通过o-脱甲基化体转化为o-去甲基曲马多(m1),通过n-脱甲基化转化为n-去甲基曲马多(m2)和n,n-二去甲基曲马多(m3),另外,通过它们的组合转化为o,n,n-三去甲基曲马多(m4)、o,n-二去甲基曲马多(m5)。m1、m4和m5进而通过第二相反应而实现葡糖醛酸结合或硫酸结合,成为直至m6~m11的缀合体。其中,m1和m1缀合体、m5和m5缀合体、以及m2为主要代谢物,m3、m4和m4缀合体为微量。除这些以外还报道了m12及之后的多个代谢物。曲马多未变化体和这些代谢物主要通过肾被排泄(约90%)。需要说明的是,本技术中单独称为单曲马多时,只要没有特别声明则指未变化体。
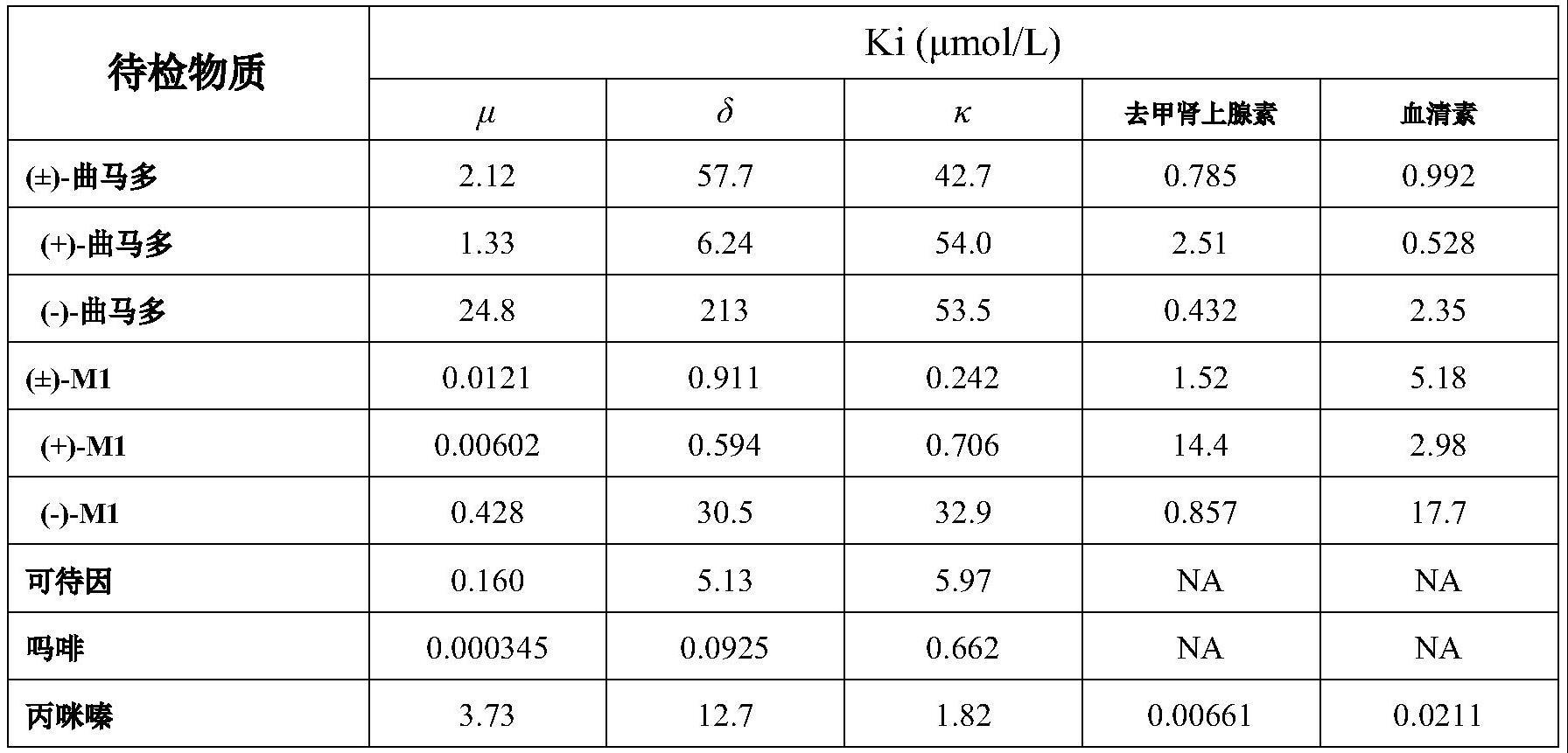
5、截止目前的研究可知,(±)-曲马多相对于μ-阿片受体的ki值仅具有吗啡的数百~数千分之一左右的亲和性。另外,与δ-和κ-阿片受体的亲和性更弱,因此,难以认为曲马多本身是通过阿片受体而发挥作用的。曲马多的镜像异构体之间,与(-)-曲马多相比,(+)-曲马多相对于μ-阿片受体的亲和性强。另一方面,曲马多代谢物中,(±)-m1相对于阿片受体的亲和性增加,特别是(+)-m1的亲和性高。作为汇总了以上那样的见解的资料,公布了以下内容(表1)。
6、[表1]
7、曲马多对阿片受体的结合亲和性和单胺再摄取抑制作用
8、
9、na:10μmol/l下无活性平均值(n=2~3)
10、(摘自持田制药株式会社制作的“tramcet(注册商标)配方片information”web网页)
11、由此可认为,曲马多对μ-阿片受体的作用的主体是作为代谢物的m1、特别是(+)-m1而并非曲马多本身,但即便如此,与吗啡、芬太尼等其它强阿片相比,已知在人类中仅有数十~数百分之一左右的结合亲和性。
12、对于具有以上那样的药代动力学、药理作用的曲马多,作为主要的副作用,已知有便秘、恶心(nausea)、呕吐、嗜睡、食欲减退、漂浮性眩晕、头痛、口渇、倦怠感等。虽然曲马多如上所述可以说相对弱,但由于是阿片的一种,这些副作用是必然出现的副作用,与吗啡等强阿片相比,认为频率低。但是,阿片的副作用的个体差异大,如上所述,曲马多还具有血清素和去甲肾上腺素的再摄入抑制作用,因此需要注意副作用可能会增强。
技术实现思路
1、发明要解决的问题
2、本发明人等在对日本一直未销售过的1日2次给药型的经口用曲马多制剂(片剂)(以下有时称为“本发明剂”。)进行临床开发的过程中发现了令人惊讶的见解,从而完成了本发明。
3、即,已经证实了本发明剂与1日4次给药型的经口剂(胶囊剂)、1日1次给药型的经口剂(片剂)(以下,将前者称为“其它制剂a”,将后者称为“其它制剂b”,有时将两者一并称为“其它制剂”。)相比副作用的出现率低。此外,进行了深入解析,结果认为,本发明剂的副作用出现率与其它制剂相比低这一效果(以下有时称为“本发明效果”。)是由给予本发明剂和其它制剂后的曲马多未变化体和m1代谢物的血药浓度等药代动力学参数的不同所带来的。
4、用于解决问题的方案
5、将含有100mg的曲马多盐酸盐的本发明剂在绝食下单次给予作为健康成人的受试者时的、受试者的血浆中的曲马多未变化体和m1的药代动力学参数与由公开资料获知的其它制剂的对应参数的比较结果如下所述(表2)。
6、[表2]
7、100mg经口单次给予时的药代动力学参数的制剂间的比较(平均值±标准偏差)
8、
9、a)生物学等效性试验、申请制剂的饮食影响试验
10、b)药物访谈表(其它制剂b)
11、c)药物访谈表(其它制剂a)
12、若观察表2的结果,可知:本发明剂的未变化体的血药浓度峰值(cmax)是处于其它制剂a的血药浓度峰值与其它制剂b的血药浓度峰值之间的中间值。另一方面,特征在于:血药浓度达峰时间(tmax)比其它制剂a和其它制剂b的血药浓度达峰时间小,血浆消除半衰期(t1/2)比其它制剂a和其它制剂b的血浆消除半衰期大。从0小时至无限大时间的血药浓度-时间曲线下面积(auc0-inf)没有大的不同。
13、另一方面,对于m1代谢物而言,本发明剂的m1的血药浓度峰值(cmax)为处于其它制剂a的血药浓度峰值与其它制剂b的血药浓度峰值之间的中间值,血药浓度达峰时间(tmax)比其它制剂a和其它制剂b的血药浓度达峰时间小,血浆消除半衰期(t1/2)比其它制剂a和其它制剂b的血浆消除半衰期大,在这点上与未变化体同样。
14、接着,给予本发明剂的1日量(100mg×2次)后的血药浓度与由公开资料获知的给予其它制剂a(50mg×4次)和其它制剂b(200mg×1次)后的血药浓度的比较结果如下所述。(表3)
15、[表3]
16、1日量的血药浓度的制剂间比较(平均值±标准偏差)
17、
18、a)本发明剂
19、b)药物访谈表(其它制剂b)
20、由表3的结果可知:给予1日量时,本发明剂的m1代谢物的血药浓度峰值(cmax)与其它制剂相比高21~29%。另外可知:对于血药浓度-时间曲线下面积(auc0-inf)而言,本发明剂与其它制剂相比高23%。
21、需要说明的是,证实了:即使在反复给予本发明剂、其它制剂的情况下,也可以得到与上述给予1日量时相同的数据。
22、原本,由于m1代谢物如上所述对μ-阿片受体具有激动作用,预测其浓度高时阿片特有的副作用出现频率也会提高,但是申请人进行的临床试验结果与其相反。
23、即,令人惊讶地,本发明剂和公开的其它制剂在临床试验中的副作用出现率的比较结果如下所述。这些副作用出现率是在该各临床试验期间观察到的数据的汇总。(表4)
24、[表4]
25、
26、得到如表4那样的结果的理由不清楚,但上述的副作用是使用阿片通常被观察到的副作用,因此本发明剂和其它制剂中的该副作用出现率的差异显然不是由两种制剂在制造上所使用的添加剂的不同所导致的。可确定的是存在如下事实:在单次或多次给药时,如果曲马多的未变化体、m1代谢物显示出上述那样的药代动力学(以下有时称为“本发明药代动力学”。),则会观察到上述那样的副作用出现频率降低。
27、上述的各临床试验中,受试人数(病例数)也较多,因此得到的副作用出现率的数据可靠性高。
28、发明人等鉴于这样的事实,完成了本发明。即,本发明如下所示,但不受这些任何限定。
29、(1)一种制剂,其特征在于,其为含有曲马多或其药学上可接受的盐作为有效成分的经口给予制剂,对人经口单次给予相当于100mg该有效成分的量时,经该经口给予的人体内的曲马多的血药浓度峰值(cmax)为150~250ng/ml。
30、(2)根据上述(1)所述的制剂,其中,前述曲马多的血药浓度达峰时间(tmax)为1~1.4小时。
31、(3)根据上述(1)或(2)所述的制剂,其中,前述曲马多的血浆消除半衰期(t1/2)为6.5~9.5小时。
32、(4)根据前述(1)~(3)中任一项所述的制剂,其中,前述曲马多的从0小时至无限大时间的血药浓度-时间曲线下面积(auc0-inf)为1800~3000ng·小时/ml。
33、(5)一种制剂,其特征在于,其为含有曲马多或其药学上可接受的盐作为有效成分的经口给予制剂,对人经口单次给予相当于100mg该有效成分的量时,经该经口给予的人体内的活性代谢物o-去甲基曲马多(m1)的血药浓度峰值(cmax)为40~65ng/ml。
34、(6)根据前述(5)所述的制剂,其中,前述m1的血药浓度达峰时间(tmax)为1.5小时以上且小于2小时。
35、(7)根据前述(5)或(6)所述的制剂,其中,前述m1的血浆消除半衰期(t1/2)为7.5~12小时。
36、(8)根据前述(5)~(7)中任一项所述的制剂,其中,前述m1的从0小时至无限大时间的血药浓度-时间曲线下面积(auc0-inf)为650~1000ng·小时/ml。
37、(9)根据前述(1)至(8)中任一项所述的制剂,其中,有效成分为曲马多盐酸盐。
38、(10)根据前述(1)~(9)中任一项所述的制剂,其为1日2次给药型。
39、(11)根据前述(10)所述的制剂,其为片剂。
40、(12)根据前述(11)所述的制剂,其为具有速释部和缓释部的双层片。
41、(13)根据前述(1)~(12)中任一项所述的制剂,其中,在基于日本药典中的一般试验法中的溶出试验法的第2法(桨法)的溶出试验中,在液温37℃下使用试验液900ml以每分钟50转进行溶出试验时,有效成分的溶出率在15分钟后为30~50重量%、在1小时后为40~60重量%、在2小时后为50~70重量%、在4小时后为60~80重量%、在6小时后为70~90重量%。
42、(14)根据前述(13)所述的制剂,其中,有效成分的溶出率在15分钟后为35~45重量%、在1小时后为45~55重量%、在2小时后为55~65重量%、在4小时后为65~75重量%、在6小时后为75~85重量%。
43、(15)一种制剂,其特征在于,为含有曲马多或其药学上可接受的盐作为有效成分的经口给予制剂,1日2次对人经口给予相当于100mg该有效成分的量时,经该经口给予的人体内的活性代谢物o-去甲基曲马多(m1)的血药浓度峰值(cmax)为65ng/ml以上。
44、(16)根据前述(15)所述的制剂,其特征在于,前述m1的cmax为65~85ng/ml。
45、(17)根据前述(15)或(16)所述的制剂,其中,前述m1的从0小时至无限大时间的血药浓度-时间曲线下面积(auc0-inf)为1500ng·小时/ml以上。
46、(18)根据(15)~(17)中任一项所述的制剂,其中,前述m1的auc0-inf为1500~1750ng·小时/ml。
47、(19)根据前述(15)~(18)中任一项所述的制剂,其中,有效成分为曲马多盐酸盐。
48、(20)根据前述(15)~(19)中任一项所述的制剂,其为1日2次给药型。
49、(21)根据前述(20)所述的制剂,其为片剂。
50、(22)根据前述(21)所述的制剂,其为具有速释部和缓释部的双层片。
51、(23)根据前述(15)~(22)中任一项所述的制剂,其中,在基于日本药典中的一般试验法中的溶出试验法的第2法(桨法)的溶出试验中,在液温37℃下使用试验液900ml以每分钟50转进行溶出试验时,有效成分的溶出率在15分钟后为30~50重量%、在1小时后为40~60重量%、在2小时后为50~70重量%、在4小时后为60~80重量%、在6小时后为70~90重量%。
52、(24)根据前述(23)所述的制剂,其中,有效成分的溶出率在15分钟后为35~45重量%、在1小时后为45~55重量%、在2小时后为55~65重量%、在4小时后为65~75重量%、在6小时后为75~85重量%。
53、发明的效果
54、本发明的制剂(本发明剂)具有与现有的含有曲马多的经口制剂相比副作用降低的效果(本发明效果)。
55、本发明效果并不是基于在作为有效成分的曲马多中以何种比率配混了何种添加剂而发挥出的效果,认为只要是与上述的本发明剂显示出同样的血药浓度等药代动力学的含有曲马多的经口制剂,则无论是何种处方均发挥同样的效果。从这样的意义上讲,本技术发明只要是以曲马多作为有效成分的经口制剂,其处方就没有任何限定。