治疗囊性纤维化的方法与流程
治疗囊性纤维化的方法
1.本技术要求于2020年12月10日提交的美国临时申请第63/123,928号;于2020年12月11日提交的美国临时申请第63/124,575号;以及于2021年2月17日提交的美国临时申请第63/150,434号的优先权权益,所述美国临时申请中的每个美国临时申请的内容通过引用相应整体并入本文。
2.本发明提供了用于治疗囊性纤维化的药物组合物和方法。
3.囊性纤维化(cf)是一种隐性遗传疾病,在全球范围内有约83,000名儿童及成人受到影响。尽管cf的治疗取得一定进展,但仍无法治愈。
4.在cf患者中,呼吸道上皮中内源性表达的cftr突变导致顶端阴离子分泌减少,使得离子和液体转运失衡。由此引起的阴离子转运减少导致肺中粘液累积增加,并且伴随微生物感染,最终导致cf患者死亡。除呼吸系统疾病之外,cf患者通常患有胃肠道问题和胰腺功能不全,如果不治疗,则会导致死亡。另外,大多数患有囊性纤维化的男性不育,并且患有囊性纤维化的女性的生育力降低。
5.关于cftr基因的序列分析已揭露出多种引起疾病的突变(cutting,g.r.等人(1990)nature 346:366-369;dean,m.等人(1990)cell 61:863:870;以及kerem,b-s.等人(1989)science 245:1073-1080;kerem,b-s等人(1990)proc.natl.acad.sci.usa 87:8447-8451)。迄今为止,已鉴定出cf基因中超过2000个突变。cf突变在位于http://www.genet.sickkids.on.ca/app的
″
囊性纤维化突变数据库
″
中列出,所述数据库通过引用整体并入本文。最普遍的致病突变是cftr氨基酸序列508位的苯丙氨酸缺失,并且通常称为f508del突变。该突变出现在大约90%的囊性纤维化病例中,并且与严重疾病相关。
6.cftr中残基508的缺失阻止新生蛋白质正确地折叠。这导致突变蛋白无法离开内质网(er)并运输至质膜。因此,存在于膜中的用于阴离子转运的cftr通道的数目远低于在表达野生型cftr,即不合突变的cftr的细胞中观察到的数目。除了运输受损之外,突变导致缺陷性通道门控。膜中通道数目的减少和门控缺陷一起导致阴离子和流体跨上皮转运减少。(quinton,p.m.(1990),faseb j.4:2709-2727)。由于f508del突变而存在缺陷的通道仍是功能性的,尽管其功能小于野生型cftr通道。(dalemans等人(1991),nature lond.354:526-528;pasyk和foskett(1995),j.cell.biochem.270:12347-50)。除f508del之外,cftr中导致缺陷性运输、合成和/或通道门控的其它致病突变可以上调或下调以改变阴离子分泌并改变疾病进展和/或严重程度。
7.cftr是在包括吸收性和分泌性上皮细胞的各种细胞类型中表达的camp/atp介导的阴离子通道,其中它调控跨膜阴离子通量以及其它离子通道和蛋白质的活性。在上皮细胞中,cftr的正常功能对于维持电解质在全身(包括呼吸和消化组织)的转运至关重要。cftr由编码蛋白质的1480个氨基酸构成,所述蛋白质由跨膜结构域的串联重复序列组成,每个跨膜结构域含有六个跨膜螺旋和一个核苷酸结合结构域。两个跨膜结构域通过大的极性调控(r)结构域与多个磷酸化位点连接,从而调控通道活性和细胞运输。
8.氯离子转运提供存在于顶端膜上的enac(上皮钠通道)和cftr以及细胞基底外侧表面上表达的na
+-k
+-atp酶泵和cl-通道的协调活性进行。氯离子从内腔侧的次级主动转运
导致细胞内氯离子积累,接着其可经由cl-通道被动地离开细胞,从而导致媒介物转运。na
+
/2cl-/k
+
共转运蛋白、na
+-k
+-atp酶泵和基底外侧表面上的基底外侧膜k
+
通道以及在内腔侧上cftr的布置协调氯离子分泌。由于水自身可能无法主动转运,因此其跨上皮的流动取决于由钠和氯离子大量流动产生的微小跨上皮渗透梯度。
9.最近已鉴定出多种cftr调节性化合物。然而,仍需要可治疗囊性纤维化和其它cftr介导的疾病(特别是这些疾病的更严重形式)或减轻其严重程度的化合物。
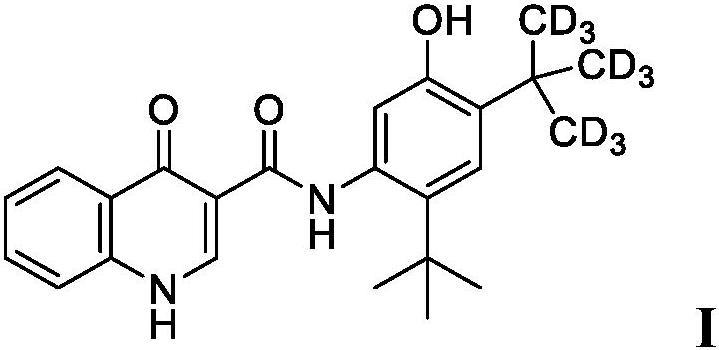
10.因此,本公开的一方面提供了包括250mg的cftr增效剂化合物、n-(2-(叔丁基)-5-羟基-4-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基)-4-氧代-1,4-二氢喹啉-3-甲酰胺(化合物i)或等效量的其药学上可接受的盐的药物组合物。化合物i可以被描绘为具有以下结构:
[0011][0012]
已经发现,与150mg q12h的依伐卡托(ivacaftor)(即,以12小时间隔每天两次)和150mg qd的化合物i(即,每天一次)相比,每天施用250mg的化合物i可以改善如通过汗氯化物(swcl)测量的治疗谱。
[0013]
本公开的其它方面提供了包括250mg的化合物i或等效量的其药学上可接受的盐的药物组合物,所述药物组合物进一步包含至少一种另外的活性药物成分和/或至少一种载体。本公开的又其它方面是治疗cftr介导的疾病囊性纤维化的方法,所述方法包括向有需要的受试者施用化合物i或等效量的其药学上可接受的盐,任选地作为包括至少一种另外的组分的药物组合物的一部分。在一些实施例中,本发明的药物组合物包括250mg的化合物i(或等效量的其药学上可接受的盐)、21.24mg的化合物ii、钙盐水合物形式d和100mg的化合物iii(或等效量的其药学上可接受的盐)。在一些实施例中,本发明的药物组合物包括125mg的化合物i(或等效量的其药学上可接受的盐)、10.62mg的化合物ii、钙盐水合物形式d和50mg的化合物iii(或等效量的其药学上可接受的盐)。
[0014]
一个实施例提供了治疗cftr介导的疾病囊性纤维化的方法,所述方法包括单独施用250mg的n-(2-(叔丁基)-5-羟基-4-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基)-4-氧代-1,4-二氢喹啉-3-甲酰胺(化合物i)或与21.24mg的(14s)-8-[3-(2-{二螺[2.0.2.1]庚-7-基}乙氧基)-1h-吡唑-1-基]-12,12-二甲基-2λ6-硫杂-3,9,11,18,23-五氮杂四环[17.3.1.111,14.05,10]二十四碳-1(22),5,7,9,19(23),20-己烯-2,2,4-酮(化合物ii)钙盐水合物形式d和/或50-100mg的(r)-1-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)-n-(1-(2,3-二羟丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1h-吲哚-5-基)环丙烷甲酰胺(化合物iii)组合施用。
[0015]
在一些实施例中,250mg的化合物i(或等效量的其药学上可接受的盐)以与20mg的化合物ii钙盐水合物形式d相同的组合物施用。在一些实施例中,250mg的化合物i以与21.24mg的化合物ii钙盐水合物形式d和100mg的化合物iii(或等效量的其药学上可接受的盐)相同的组合物施用。在一些实施例中,包括250mg的化合物i(或等效量的其药学上可接
受的盐)的组合物与包括21.24mg的化合物ii钙盐水合物形式d和/或100mg的化合物iii(或等效量的其药学上可接受的盐)的单独的组合物共施用。在一些实施例中,250mg的化合物i以与21.24mg的化合物ii钙盐水合物形式d和100mg的化合物iii(或等效量的其药学上可接受的盐)相同的组合物施用。在一些实施例中,250mg的化合物i、21.24mg的化合物ii钙盐水合物形式d和100mg的化合物iii(或等效量的其药学上可接受的盐)以两种等效剂量组合物每天施用一次。
附图说明
[0016]
图1提供了结晶化合物i(游离形式)形式a的xrpd图案。
[0017]
图2示出了结晶化合物i(游离形式)形式a的
13
c固态nmr谱。
[0018]
图3提供了结晶化合物i钙盐水合物形式a的xrpd图案。
[0019]
图4示出了化合物i钙盐水合物形式a的
13
c固态nmr谱。
[0020]
图5提供了结晶化合物i钙盐水合物形式d的xrpd图案。
[0021]
图6示出了化合物i钙盐水合物形式d的
13
c固态nmr谱。
[0022]
定义
[0023]
如本公开全文中使用的
″
化合物i
″
是指n-(2-(叔丁基)-5-羟基-4-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基)-4-氧代-1,4-二氢喹啉-3-甲酰胺,其可以被描绘为具有以下结构:
[0024][0025]
化合物i可以呈药学上可接受的盐的形式。化合物i和其药学上可接受的盐先前已在美国专利第8,865,902号、第9,181,192号和第9,512,079号以及国际专利公开第wo 2012/158885号、第wo 2014/078842号、第wo 2017/053455号和第wo 2018/080591号中有所描述,所述专利中的每一个通过引用并入本文。
[0026]
在一些实施例中,化合物i中的每个氘的同位素富集因子可以变化。术语
″
同位素富集因子
″
是指指定同位素之同位素丰度与天然丰度之间的比率。在一些实施例中,化合物i中的每个指定氘原子的同位素富集因子为至少3500(在每个指定氘原子处的52.5%氘并入量)、至少4000(60%氘并入量)、至少4500(67.5%氘并入量)、至少5000(75%氘并入量)、至少5500(82.5%氘并入量)、至少6000(90%氘并入量)、至少6333.3(95%氘并入量)、至少6466.7(97%氘并入量)、至少6600(99%氘并入量)或至少6633.3(99.5%氘并入量)。
[0027]
如本文所使用的
″
化合物ii
″
是指(14s)-8-[3-(2-{二螺[2.0.2.1]庚-7-基}乙氧基)-1h-吡唑-1-基]-12,12-二甲基-2λ6-硫杂-3,9,11,18,23-五氮杂四环[17.3.1.111,14.05,10]二十四碳-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮,其可以用以下结构描绘:
[0028][0029]
化合物ii和氘化衍生物和其药学上可接受的盐首先在国际专利公开wo 2019/161078(通过引用并入本文)中描述。
[0030]
在一些实施例中,化合物ii呈钙盐水合物形式d的形式。20mg的化合物ii相当于21.24mg的化合物ii钙盐水合物形式d。在一些实施例中,化合物ii钙盐水合物形式d的特征在于x射线粉末衍射图,所述x射线粉末衍射图具有在以下各处的信号:6.1
±
0.2度2θ、16.2
±
0.2度2θ和22.8
±
0.2度2θ。在一些实施例中,化合物ii钙盐水合物形式d的特征在于x射线粉末衍射图,所述x射线粉末衍射图具有:(a)在6.1
±
0.2度2θ、16.2
±
0.2度2θ和22.8
±
0.2度2θ处的信号;以及(b)选自5.5
±
0.2度2θ、15.5
±
0.2度2θ、19.7
±
0.2度2θ、21.5
±
0.2度2θ、22.1
±
0.2度2θ、23.0
±
0.2度2θ和27.6
±
0.2度2θ的一个或多个信号。
[0031]
在一些实施例中,化合物ii钙盐水合物形式d的特征在于x射线粉末衍射图,所述x射线粉末衍射图具有:(a)在6.1
±
0.2度2θ、16.2
±
0.2度2θ和22.8
±
0.2度2θ处的信号;以及(b)选自5.5
±
0.2度2θ、15.5
±
0.2度2θ、19.7
±
0.2度2θ、21.5
±
0.2度2θ、22.1
±
0.2度2θ、23.0
±
0.2度2θ和27.6
±
0.2度2θ的两个或更多个信号。在一些实施例中,化合物ii钙盐水合物形式d的特征在于x射线粉末衍射图,所述x射线粉末衍射图具有:(a)在6.1
±
0.2度2θ、16.2
±
0.2度2θ和22.8
±
0.2度2θ处的信号;以及(b)选自5.5
±
0.2度2θ、15.5
±
0.2度2θ、19.7
±
0.2度2θ、21.5
±
0.2度2θ、22.1
±
0.2度2θ、23.0
±
0.2度2θ和27.6
±
0.2度2θ的三个或更多个信号。在一些实施例中,化合物ii钙盐水合物形式d的特征在于x射线粉末衍射图,所述x射线粉末衍射图具有:(a)在6.1
±
0.2度2θ、16.2
±
0.2度2θ和22.8
±
0.2度2θ处的信号;以及(b)选自5.5
±
0.2度2θ、15.5
±
0.2度2θ、19.7
±
0.2度2θ、21.5
±
0.2度2θ、22.1
±
0.2度2θ、23.0
±
0.2度2θ和27.6
±
0.2度2θ的四个或更多个信号。
[0032]
在一些实施例中,化合物ii钙盐水合物形式d的特征在于x射线粉末衍射图,所述x射线粉末衍射图具有在以下各处的信号:6.1
±
0.2度2θ、16.2
±
0.2度2θ和22.8
±
0.2度2θ以及27.6
±
0.2度2θ。在一些实施例中,化合物ii钙盐水合物形式d的特征在于x射线粉末衍射图,所述x射线粉末衍射图具有在以下各处的信号:6.1
±
0.2度2θ、15.5
±
0.2度2θ、16.2
±
0.2度2θ、19.7
±
0.2度2θ、22.8
±
0.2度2θ和27.6
±
0.2度2θ。在一些实施例中,化合物ii钙盐水合物形式d的特征在于基本上类似于图5的x射线粉末衍射图。
[0033]
在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有选自以下的一个或多个峰的
13
c固态核磁共振(
13
c ssnmr)谱:179.8
±
0.2ppm、130.2
±
0.2ppm、125.6
±
0.2ppm、120.9
±
0.2ppm、55.2
±
0.2ppm、44.3
±
0.2ppm、35.0
±
0.2ppm和1.6
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有选自以下的两个或更多个峰的
13
c ssnmr谱:179.8
±
0.2ppm、130.2
±
0.2ppm、125.6
±
0.2ppm、120.9
±
0.2ppm、55.2
±
0.2ppm、44.3
±
0.2ppm、35.0
±
0.2ppm和1.6
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有选自以下的三个或更多个峰的
13
c ssnmr谱:179.8
±
0.2ppm、
130.2
±
0.2ppm、125.6
±
0.2ppm、120.9
±
0.2ppm、55.2
±
0.2ppm、44.3
±
0.2ppm、35.0
±
0.2ppm和1.6
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有选自以下的四个或更多个峰的
13
c ssnmr谱:179.8
±
0.2ppm、130.2
±
0.2ppm、125.6
±
0.2ppm、120.9
±
0.2ppm、55.2
±
0.2ppm、44.3
±
0.2ppm、35.0
±
0.2ppm和1.6
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有选自以下的五个或更多个峰的
13
c ssnmr谱:179.8
±
0.2ppm、130.2
±
0.2ppm、125.6
±
0.2ppm、120.9
±
0.2ppm、55.2
±
0.2ppm、44.3
±
0.2ppm、35.0
±
0.2ppm和1.6
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有选自以下的六个或更多个峰的
13
c ssnmr谱:179.8
±
0.2ppm、130.2
±
0.2ppm、125.6
±
0.2ppm、120.9
±
0.2ppm、55.2
±
0.2ppm、44.3
±
0.2ppm、35.0
±
0.2ppm和1.6
±
0.2ppm。
[0034]
在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有选自以下的一个或多个峰的
13
c ssnmr谱:130.2
±
0.2ppm、125.6
±
0.2ppm和35.0
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有选自以下的两个或更多个峰的
13
c ssnmr谱:130.2
±
0.2ppm、125.6
±
0.2ppm和35.0
±
0.2ppm。在一些实施例中,化合物i钙盐水合物形式d的特征为具有含有在以下各处的峰的
13
c ssnmr谱:130.2
±
0.2ppm、125.6
±
0.2ppm和35.0
±
0.2ppm。
[0035]
在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有在以下各处的峰的
13
c ssnmr谱:(a)130.2
±
0.2ppm、125.6
±
0.2ppm和/或35.0
±
0.2ppm;以及(b)176.9
±
0.2ppm、160.9
±
0.2ppm、142.0
±
0.2ppm和/或98.6
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有在以下各处的峰的
13
c ssnmr谱:(a)130.2
±
0.2ppm、125.6
±
0.2ppm和/或35.0
±
0.2ppm;以及(b)176.9
±
0.2ppm、160.9
±
0.2ppm、142.0
±
0.2ppm和98.6
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有在以下各处的峰的
13
c ssnmr谱:(a)130.2
±
0.2ppm、125.6
±
0.2ppm和35.0
±
0.2ppm;以及(b)176.9
±
0.2ppm、160.9
±
0.2ppm、142.0
±
0.2ppm和/或98.6
±
0.2ppm。在一些实施例中,化合物ii钙盐水合物形式d的特征为具有含有在以下各处的峰的
13
c ssnmr谱:130.2
±
0.2ppm、125.6
±
0.2ppm、35.0
±
0.2ppm、176.9
±
0.2ppm、160.9
±
0.2ppm、142.0
±
0.2ppm和98.6
±
0.2ppm。
[0036]
在一些实施例中,化合物ii钙盐水合物形式d的特征在于基本上类似于图6的
13
c ssnmr谱。
[0037]
在一些实施例中,化合物ii钙盐水合物形式d的特征在于三斜晶系、p1空间群和在配备有cu k
α
辐射和互补型金属氧化物半导体(cmos)检测器的bruker衍射仪上在100k下测量的以下晶胞尺寸:
[0038][0039]
如本公开全文中使用的
″
化合物iii
″
是指(r)-1-(2,2-二氟苯并[d][1,3]二氧杂环戊烯-5-基)-n-(1-(2,3-二羟丙基)-6-氟-2-(1-羟基-2-甲基丙-2-基)-1h-吲哚-5-基)环丙烷甲酰胺:
[0040][0041]
在一些实施例中,化合物iii呈药学上可接受的盐的形式。化合物iii和其药学上可接受的盐先前在国际专利公开wo 2010/053471(通过引用并入本文)中公开。
[0042]
如本文所用,
″
cftr
″
是指囊性纤维化跨膜传导调控因子。
[0043]
如本文所使用的,
″
突变
″
可以指cftr基因或cftr蛋白中的突变。
″
cftr基因突变
″
是指cftr基因中的突变,并且
″
cftr蛋白突变
″
是指cftr蛋白中的突变。基因缺陷或突变或基因中的核苷酸的变化通常产生从此基因翻译的cftr蛋白的突变或框移。
[0044]
术语
″
f508del
″
是指在位置508处缺乏氨基酸苯丙氨酸的突变体cftr蛋白。
[0045]
如本文所使用的,对于特定基因突变为
″
纯合
″
的患者在每个等位基因上具有相同的突变。
[0046]
如本文所使用的,对于特定基因突变为
″
杂合
″
的患者在一个等位基因上具有特定突变,并且在另一个等位基因上具有不同的突变。
[0047]
如本文所使用的,术语
″
调节剂
″
是指增加如蛋白质等生物化合物活性的化合物。例如,cftr调节剂是增加cftr活性的化合物。由cftr调节剂引起的活性增加包括但不限于对cftr进行校正、增强、稳定化和/或扩增的化合物。
[0048]
如本文所用,术语
″
cftr校正剂
″
是指促进cftr的加工和运输以增加细胞表面处cftr的量的化合物。本文所公开的化合物ii和iii是cftr校正剂。
[0049]
如本文所用,术语
″
cftr增效剂
″
是指增加位于细胞表面处的cftr蛋白的通道活性,从而导致离子转运增强的化合物。本文所公开的化合物i是cftr增效剂。
[0050]
如本文所使用的,术语
″
活性药物成分
″
或
″
治疗剂
″
(
″
api
″
)是指生物活性化合物。
[0051]
如本文所用,术语
″
药学上可接受的盐
″
是指本公开的化合物的盐形式,其中盐是无毒的。本公开的化合物的药学上可接受的盐包括衍生自合适的无机和有机酸和碱的那些盐。药学上可接受的盐是本领域公知的。例如,s.m.berge等人在《药物科学杂志(j.pharmaceutical sciences)》,1977,66,1-19中详细描述了药学上可接受的盐。
[0052]
如本文所用,术语
″
非晶形
″
是指在其分子位置中不具有长程有序的固体材料。非晶形固体通常是其中分子以随机方式排列,使得不存在明确的排列(例如分子堆积)并且不存在长程有序的过冷液体。非晶形固体通常是各向同性的,即在所有方向上表现出类似的性质并且不具有确定的熔点。例如,非晶形材料是在其x射线粉末衍射(xrpd)图案中不具有尖锐特征结晶峰的固体材料(即,如通过xrpd所确定不是结晶的)。相反,在其xrpd图案中出现一个或多个宽峰(例如,晕)。宽峰是非晶形固体的特征。有关非晶形材料和结晶材料的xrpd的比较,参见us2004/0006237。在一些实施例中,固体材料可以包括非晶形化合物,并且所述材料的特征可以例如在于在其xrpd谱中缺少尖锐特征结晶峰(即所述材料不是结晶,而是非晶形,如通过xrpd所确的)。相反,在材料的xrpd图案中可以出现一个或多个宽峰(例如,晕)。有关非晶形材料和结晶材料的xrpd的比较,参见us2004/0006237。与纯结晶固体的熔融范围相比,包含非晶形化合物的固体材料的特征可以(例如)在于该固体材料的熔
融温度范围更宽。其它技术,例如拉曼光谱、红外光谱和固态nmr可以用于表征结晶或非晶形形式。
[0053]
在一些实施例中,固体材料可以包含结晶固体和非晶形固体的混合物。制备成包括非晶形化合物的固体材料也可以例如含有至多30%的结晶固体。在一些实施例中,制备成包含非晶形化合物的固体材料也可以例如含有至多25%、20%、15%、10%、5%或2%的结晶固体。在其中固体材料含有结晶固体与非晶形固体的混合物的实施例中,诸如xrpd等表征数据可含有结晶固体与非晶形固体两者的指示物。
[0054]
如本文所用,术语
″
基本上非晶形
″
是指在其分子的位置中具有很少或没有长程有序的固体材料。例如,基本上非晶形材料具有小于15%的结晶度(例如,小于10%的结晶度、小于5%的结晶度或小于2%的结晶度)。还应注意,术语
′
基本上非晶形
′
包含描述词
′
非晶形
′
,其指不具有(0%)结晶度的材料。
[0055]
如本文所用,术语
″
基本上结晶的
″
是指几乎不具有或不具有非晶形分子的固体材料。例如,基本上结晶的材料具有小于15%的非晶形分子(例如,小于10%的非晶形分子、小于5%的非晶形分子或小于2%的非晶形分子)。还应注意,术语
″
基本上结晶的
″
包括描述词
″
结晶的
″
,其指为100%结晶形式的材料。
[0056]
如本文所用,术语
″
xrpd
″
是指x射线粉末衍射的分析表征方法。在环境条件下使用衍射仪以透射或反射几何学记录本文所公开的xrpd图案。
[0057]
如本文所用,术语
″
环境条件
″
意指室温、露天条件和不受控制的湿度条件。术语
″
室温
″
和
″
环境温度
″
意指15℃至30℃。
[0058]
如本文所用,术语
″
x射线粉末衍射图
″
、
″
x射线粉末衍射图案
″
、
″
xrpd图案
″
、
″
xrpd谱
″
可互换地是指实验获得的绘制信号位置(在横坐标上)对信号强度(在纵坐标上)的图案。对于非晶形材料,x射线粉末衍射图可包括一个或多个宽信号;并且对于结晶材料,x射线粉末衍射图可包括一个或多个信号,每一个信号由其以度2θ(
°
2θ)为单位量度的角度值鉴别,绘示在x射线粉末衍射图的横坐标上,其可表示为
″
在......
°
2θ处的信号
″
、
″
在......的2θ值处的信号
″
和/或
″
在至少......个选自......的2θ值处的信号
″
。
[0059]
如本文所用,
″
信号
″
或
″
峰
″
是指xrpd图案中以计数测量的强度处于局部最大值的点。本领域的普通技术人员将认识到,xrpd图案中的一个或多个信号(或峰)可重叠,并且可例如对肉眼不明显。实际上,本领域的普通技术人员将认识到,一些业内认可的方法能够并且适用于确定信号是否存在于图案中,诸如rietveld精修。
[0060]
如本文所用,
″
在......
°
2θ处的信号
″
是指如在x射线粉末衍射实验中所测量并观察到的x射线反射位置(
°
2θ)。
[0061]
所测量角度值的可重复性在
±
0.2
°
2θ范围内,即角度值可在所列举的角度值+0.2
°
2θ处、角度值-0.2
°
2θ处或在该等两个终点(角度值+0.2
°
2θ和角度值-0.2
°
2θ)之间的任一值处。
[0062]
术语
″
信号强度
″
和
″
峰强度
″
可互换地指在给定x射线粉末衍射图内的相对信号强度。可影响相对信号或峰强度的因素包括样品厚度和优选定向(例如结晶颗粒不随机分布)。
[0063]
如本文所用,当两个衍射图中至少90%(诸如至少95%、至少98%或至少99%)的信号重叠时,x射线粉末衍射图
″
基本上与[特定]图中类似
″
。在确定
″
基本类似性
″
时,本领
域的普通技术人员应理解,即使对于相同的结晶形式,xrpd衍射图中的强度和/或信号位置也可存在变化。因此,本领域的普通技术人员将理解,xrpd衍射图中的信号最大值(以
°
2θ为单位)通常意味着该值鉴别为所报告值
±
0.2
°
2θ,其为业内公认的方差。
[0064]
如本文所使用的,当两个谱中的至少90%,如至少95%、至少98%或至少99%的信号重叠时,
13
c ssnmr谱
″
基本上与[特定]图中的谱类似
″
。在确定
″
基本类似性
″
时,本领域的普通技术人员应理解,即使对于相同的结晶形式,ssnmr谱中的强度和/或信号位置也可存在变化。因此,本领域的普通技术人员应理解,ssnmr谱中的化学位移(以本文中所提及的百万分率(ppm)为单位)通常意味着该值鉴别为所报告值
±
0.2ppm,其为业内公认的方差。
[0065]
如本文所用的术语
″
具有在......2θ值处的信号的x射线粉末衍射图
″
是指含有如在x射线粉末衍射实验中所测量并观察到的x射线反射位置(
°
2θ)的xrpd图案。
[0066]
如本文所用,术语
″
dsc
″
是指差示扫描量热法的分析方法。
[0067]
如本文所使用,术语
″
溶剂
″
是指可至少部分溶解产物(产物溶解度>1g/l)的任何液体。
[0068]
如本文所用,术语
″
分散体
″
是指其中一种物质(即分散相)以离散单元分布于整个第二物质(连续相或媒介物)中的分散系统。分散相的大小可以显著变化(例如,纳米尺寸的胶体颗粒,到几微米大小)。通常,分散相可以是固体、液体或气体。在固体分散体的情况下,分散相和连续相都是固体。在药物应用中,固体分散体可包括非晶形聚合物(连续相)中的结晶药物(分散相);或替代地,非晶形聚合物(连续相)中的非晶形药物(分散相)。在一些实施例中,固体分散体包括构成分散相的聚合物和构成连续相的药物。或者,固体分散体包括构成分散相的药物和构成连续相的聚合物。
[0069]
术语
″
患者
″
和
″
受试者
″
可互换使用并且指包括人的动物。
[0070]
如本文所使用,术语
″
治疗(treatment/treating)
″
等通常意指改善受试者的cf或其症状或减轻cf或cf的症状中的一种或多种的严重程度。如本文所用,
″
治疗
″
包括但不限于以下:受试者的生长增加、体重增益增加、肺粘液减少、胰腺和/或肝功能改善、胸部感染减轻和/或咳嗽或呼吸急促减轻。可以根据本领域已知的标准方法和技术容易地评估这些症状中任一者的改善或其严重程度的减轻。
[0071]
如本文所使用,当提及两种或更多种化合物、药剂或另外的药物活性成分时,术语
″
与......组合
″
意指在彼此之前、与其同时或在其之后向患者施用两种或更多种化合物、药剂或药物活性成分。
[0072]
当与组合物或剂型的成分的剂量、量或重量百分比结合使用时,术语
″
约
″
和
″
大约
″
包括包括本领域的普通技术人员认为可提供与自指定剂量、量或重量百分比所获得的药理学作用等效的药理学作用的指定剂量、量或重量百分比的值、或该剂量、量或重量百分比的范围。术语
″
约
″
和
″
大约
″
可以指由本领域技术人员确定的特定值的可接受误差,其部分取决于如何测量或确定所述值。在一些实施例中,术语
″
约
″
和
″
大约
″
意指给定值或范围的20%、15%、10%、5%、4%、3%、2%、1%或0.5%以内。
[0073]
本领域的普通技术人员将认识到,当公开
″
化合物或其药学上可接受的盐
″
的量时,化合物的药学上可接受的盐形式的量是等于化合物的游离碱的浓度的量。应注意,本文所公开的化合物或其药学上可接受的盐的量是以其游离碱形式计。例如,
″
100mg的选自化合物i和其药学上可接受的盐的至少一种化合物
″
包含100mg的化合物i和相当于100mg的化
合物i的化合物i的药学上可接受的盐的浓度。
[0074]
合适的药学上可接受的盐是例如s.m.berge等人j.pharmaceutical sciences,1977,66,1-19中所公开的那些盐。例如,该文章的表1提供以下药学上可接受的盐:
[0075]
表1:
[0076][0077][0078]
药学上可接受的酸加成盐的非限制性实例包括:与无机酸形成的盐,所述无机酸诸如盐酸、氢溴酸、磷酸、硫酸或高氯酸;与有机酸形成的盐,所述有机酸诸如乙酸、草酸、马来酸、酒石酸、柠檬酸、琥珀酸或丙二酸;以及通过使用本领域中使用的其它方法诸如离子
交换形成的盐。药学上可接受的盐的非限制性实例包括己二酸盐、褐藻酸盐、抗坏血酸、天冬氨酸盐、苯磺酸盐、苯甲酸盐、硫酸氢盐、硼酸盐、丁酸盐、樟脑酸盐、樟脑磺酸盐、柠檬酸盐、环戊烷丙酸盐、二葡萄糖酸盐、十二烷基硫酸、乙烷磺酸盐、甲酸盐、富马酸盐、葡庚糖酸盐、甘油磷酸盐、葡糖酸盐、半硫酸盐、庚酸盐、己酸盐、氢碘酸盐、2-羟基-乙烷磺酸盐、乳糖醛酸盐、乳酸盐、月桂酸盐、月桂基硫酸盐、苹果酸盐、马来酸盐、丙二酸盐、甲烷磺酸盐、2-萘磺酸盐、烟酸盐、硝酸盐、油酸盐、草酸盐、棕榈酸盐、双羟萘酸盐、果胶酸盐、过硫酸盐、3-苯基丙酸盐、磷酸盐、苦味酸盐、特戊酸盐、丙酸盐、硬脂酸盐、琥珀酸盐、硫酸盐、酒石酸盐、硫氰酸盐、对甲苯磺酸盐、十一烷酸盐和戊酸盐。衍生自适当碱的药学上可接受的盐包括碱金属、碱土金属、铵和n
+
(c
1-4
烷基)4盐。本公开还设想本文公开的化合物的任何碱性含氮基团的季铵化。碱金属和碱土金属盐的合适非限制性实例包括钠、锂、钾、钙和镁。药学上可接受的盐的其它非限制性实例包括铵、季铵以及使用抗衡离子诸如卤离子、氢氧根、羧酸根、硫酸根、磷酸根、硝酸根、低碳数烷基磺酸根和芳基磺酸根形成的胺阳离子。药学上可接受的盐的其它合适的非限制性实例包括苯磺酸盐和葡糖胺盐。
[0079]
治疗方法
[0080]
cftr突变可能影响cftr的数量,即细胞表面处cftr通道的数量,或者其可以影响cftr功能,即每个通道打开和转运离子的功能。影响cftr数量的突变包括导致合成缺陷(i类缺陷)的突变、导致加工和运输缺陷(ii类缺陷)的突变、导致cftr合成减少(v类缺陷)的突变以及降低cftr的表面稳定性(vi类缺陷)的突变。影响cftr功能的突变包括导致门控缺陷(iii类缺陷)的突变和导致传导缺陷(iv类缺陷)的突变。一些cftr突变表现出多种类别的特征。
[0081]
在一些实施例中,本文公开了治疗患者的囊性纤维化的方法,所述方法包括向如人等患者施用有效量的化合物、其药学上可接受的盐或前述任一项的氘化类似物;或本公开的药物组合物,其中所述患者患有囊性纤维化。在一些实施例中,患者具有f508del/最小功能(mf)基因型、f508del/f508del基因型(针对f508del突变为同型接合的)、f508del/门控基因型,或f508del/残余功能(rf)基因型。在一些实施例中,患者是异型接合的并且具有一个f508del突变。
[0082]
如本文所用,
″
最低功能(mf)突变
″
是指与最低cftr功能相关的cftr基因突变(极低至无功能cftr蛋白),并且包括例如与cftr通道打开和关闭能力的严重缺乏相关的突变,称为缺陷型通道门控或
″
门控突变
″
;与cftr细胞加工及其向细胞表面的递送的严重缺乏相关的突变;与没有(或最少)cftr合成相关的突变;以及与通道传导的严重缺陷相关的突变。
[0083]
在一些实施例中,患者是异型接合的,并且在选自表2的一个等位基因上具有f508del突变并且在另一个等位基因上具有突变:
[0084]
表2:cftr突变
[0085]
[0086][0087]
在一些实施例中,患者是异型接合的,并且在选自表3的一个等位基因上具有f508del突变并且在另一个等位基因上具有突变。
[0088]
表3
[0089]
[0090][0091]
在一些实施例中,患者具有选自表4的至少一个突变。在一些实施例中,患者不具有f508del突变并且具有选自表4的至少一个突变。
[0092]
表4
[0093]
[0094][0095]
在一些实施例中,本公开还涉及使用上述化合物的同位素标记的衍生物的治疗方法。在一些实施例中,上述化合物或其药学可接受的盐的同位素标记的衍生物,其中一个或多个原子已被一个或多个原子取代,所述一个或多个原子具有不同于通常天然存在的(同位素标记的)原子的原子质量或质量数目的原子质量或质量数目。可商购并且适用于本公开的同位素的实例包括氢、碳、氮、氧、磷、氟和氯的同位素,分别例如2h、3h、
13
c、
14
c、
15
n、
18
o、
17
o、
31
p、
32
p、
35
s、
18
f和
36
cl。
[0096]
同位素标记的化合物和盐可以多种有益的方式使用。它们可适用于药物和/或各种类型的测定,诸如底物组织分布测定。例如,氚(3h)和/或碳-14(
14
c)标记的化合物因制备相对简单和可侦测性优良而特别适用于各种类型的测定,诸如底物组织分布测定。例如,氘(2h)标记的化合物在治疗上可用并且相较于非2h标记的化合物具有潜在的治疗优点。一般来说,相较于非同位素标记的化合物和盐,氘(2h)标记的化合物和盐可以由于以下所述的动力学同位素效应而具有较高的代谢稳定性。更高的代谢稳定性直接转化为增加的体内半衰期或较低的剂量,这可为所期望的。同位素标记的化合物和盐通常可以通过进行本文的合成方案和相关描述中、实例部分和制备部分中所公开的程序,用容易获得的同位素标记的反应物置换非同位素标记的反应物来制备。
[0097]
在一些实施例中,同位素标记的化合物和盐是氘(2h)标记的化合物和盐。在一些具体实施例中,同位素标记的化合物和盐被氘(2h)标记,其中一个或多个氢原子已被氘置
换。在化学结构中,氘表示为
″d″
。
[0098]
氘(2h)标记的化合物和盐可以通过一级动力学同位素效应来操纵化合物的氧化代谢。一级动力学同位素效应是由同位素核交换引起的化学反应速率的变化,这继而又由该同位素交换后形成共价键所必需的基态能量的变化引起。较重同位素的交换通常导致化学键的基态能量降低,并且因此导致限速键断裂的减少。如果沿着多产物反应的坐标在鞍点区域中或附近发生键断裂,则产物分布比率可以显著改变。解释:如果氘键结至在不可交换位置处的碳原子,则km/kd=2-7的速率差异是典型的。关于进一步讨论,参见s.l.harbeson和r.d.tung,药物发现和开发中的氘(deuterium in drug discovery and development),《药物化学年度报告(ann.rep.med.chem.)》2011,46,403-417,其通过引用全文并入本文。
[0099]
并入本公开的同位素标记的化合物和盐中的同位素(例如,氘)的浓度可以由同位素富集因子限定。如本文所用,术语
″
同位素富集因子
″
意指指定同位素之同位素丰度与天然丰度之间的比率。在一些实施例中,如果本公开的化合物中的取代基表示为氘,则此类化合物对于每个指定氘原子具有的同位素富集因子为至少3500(在每个指定氘原子处的52.5%氘并入量)、至少4000(60%氘并入量)、至少4500(67.5%氘并入量)、至少5000(75%氘并入量)、至少5500(82.5%氘并入量)、至少6000(90%氘并入量)、至少6333.3(95%氘并入量)、至少6466.7(97%氘并入量)、至少6600(99%氘并入量)或至少6633.3(99.5%氘并入量)。
[0100]
当发现和开发治疗剂时,本领域技术人员试图优化药代动力学参数同时保持期望的体外性质。可以合理地假设,药物动力学特性较差的许多化合物容易发生氧化代谢。
[0101]
本文所公开的一方面提供了治疗囊性纤维化和其它cftr介导的疾病的方法,所述方法包括施用250mg的化合物i(或等效量的药学上可接受的盐)。在一些实施例中,250mg的化合物i(或等效量的药学上可接受的盐)单独或与另一种cftr调节剂组合以单个剂量每天施用。在一些实施例中,250mg的化合物i(或等效量的药学上可接受的盐)作为125mg剂量单独或与另一种cftr调节剂组合每天施用一次。
[0102]
本文所公开的一方面提供了通过每天与21.24mg的化合物ii钙盐水合物形式d和100mg的化合物iii(或等效量的其药学上可接受的盐)组合施用250mg的化合物i(或等效量的其药学上可接受的盐)治疗囊性纤维化和其它cftr介导的疾病的方法。在一些实施例中,250mg的化合物i(或等效量的其药学上可接受的盐)、21.24mg的化合物ii钙盐水合物形式d和100mg的化合物iii(或等效量的其药学上可接受的盐)以单独的药物组合物每天施用。在一些实施例中,250mg的化合物i(或等效量的其药学上可接受的盐)、21.24mg的化合物ii钙盐水合物形式d和100mg的化合物iii(或等效量的其药学上可接受的盐)以单一药物组合物每天施用。在一些实施例中,250mg的化合物i(或等效量的其药学上可接受的盐)、21.24mg的化合物ii钙盐水合物形式d和100mg的化合物iii(或等效量的其药学上可接受的盐)以两种等效药物组合物一起每天施用一次。
[0103]
药物组合物
[0104]
本发明的另一方面提供了用于治疗囊性纤维化的药物组合物。在一些实施例中,本发明的药物组合物包括250mg的化合物i(或等效量的其药学上可接受的盐)。在一些实施例中,本发明的药物组合物包括125mg的化合物i(或等效量的其药学上可接受的盐)。
[0105]
在一些实施例中,本发明的药物组合物包括250mg的化合物i、21.24mg的化合物ii钙盐水合物形式d和100mg的化合物iii。在一些实施例中,本发明的药物组合物包括125mg的化合物、10.62mg的化合物ii钙盐水合物形式d和50mg的化合物iii。
[0106]
在一些实施例中,本文所公开的药物组合物(例如,片剂)包含包含化合物i的第一固体分散体(例如,喷雾干燥分散体)和包含化合物iii的第二固体分散体(例如,喷雾干燥分散体)。化合物i的非氘化类似物的固体分散体和制备此类分散体的方法公开于pct公开第wo 2007/079139号中,所述公开通过引用并入本文。这些相同的固体分散体适合于与化合物i一起使用。化合物iii的固体分散体和其制备方法公开于pct公开第wo 2011/119984号和第wo 2015/160787号中,所述公开通过引用并入本文。
[0107]
在一些实施例中,本发明的药物组合物包括:按组合物的重量计,约39.9wt%的包括化合物i的固体分散体(其中固体分散体包括按固体分散体的重量计,80wt%化合物i、19.5wt%醋酸羟丙甲纤维素琥珀酸酯和0.5wt%月桂基硫酸钠);按组合物的重量计,约2.7wt%的化合物ii钙盐水合物形式d;以及按组合物的重量计,约16.0wt%的包括化合物iii的固体分散体(其中固体分散体包括按固体分散体的重量计,80wt%化合物i和20wt%羟丙甲纤维素)。
[0108]
本领域已知的任何合适的药物组合物可以用于化合物i、化合物ii钙盐水合物形式d和化合物iii。用于化合物i和其药学上可接受的盐的一些示例性药物组合物可见于us 8,865,902、us 9,181,192、us 9,512,079、wo 2017/053455和wo 2018/080591中,所述专利中的所有专利通过引用并入本文。包括化合物ii和其药学上可接受的盐的示例性药物组合物公开于wo 2019/161078和wo 2020/102346中。用于化合物iii和其药学上可接受的盐的示例性药物组合物公开于wo 2011/119984和wo 2014/014841中,所述专利通过引用并入本文。
[0109]
本文所公开的药物组合物可以任选地进一步包括至少一种药学上可接受的载体。至少一种药学上可接受的载体可选自佐剂和媒介物。如本文所使用,至少一种药学上可接受的载体包括适合于所需的特定剂型的任何和所有溶剂、稀释剂、其它液体媒介物、分散助剂、悬浮助剂、表面活性剂、等渗剂、增稠剂、乳化剂、防腐剂、固体粘合剂和润滑剂。remington:the science and practice of pharmacy,第21版,2005,d.b.troy编辑,lippincott williams&wilkins,philadelphia和encyclopedia of pharmaceutical technology,j.swarbrick和j.c.boylan编辑,1988-1999,marcel dekker,new york公开用于配制药物组合物的各种载体和用于制备其的已知技术。除非任何常规载体与本公开的化合物不相容,诸如通过产生任何不合需要的生物作用或另外以有害方式与药物组合物的任何其它组分相互作用,否则预期其使用在本公开的范围内。合适的药学上可接受的载体的非限制性实例包括但不限于离子交换剂、氧化铝、硬脂酸铝、卵磷脂、血清蛋白(例如人血清白蛋白)、缓冲物质(例如磷酸盐、甘氨酸、山梨酸和山梨酸钾)、饱和植物脂肪酸偏甘油酯混合物、水盐和电解质(诸如硫酸鱼精蛋白、磷酸氢二钠、磷酸氢钾、氯化钠和锌盐)、胶体二氧化硅、三硅酸镁、聚乙烯吡咯烷酮、聚丙烯酸酯、蜡、聚乙烯-聚氧丙烯嵌段聚合物、羊毛脂、糖(诸如乳糖、葡萄糖和蔗糖)、淀粉(诸如玉米淀粉和马铃薯淀粉)、纤维素和其衍生物(诸如羧甲基纤维素钠、乙基纤维素和乙酸纤维素)、粉状黄蓍胶、麦芽、明胶、滑石、赋形剂(诸如可可脂和栓剂蜡)、油类(诸如花生油、棉籽油、红花油、芝麻油、橄榄油、玉米油和大豆
油)、二醇类(诸如丙二醇和聚乙二醇)、酯类(诸如油酸乙酯和月桂酸乙酯)、琼脂、缓冲剂(诸如氢氧化镁和氢氧化铝)、海藻酸、无热原质水、等渗盐水、林格氏溶液(ringer
′
s solution)、乙醇、磷酸盐缓冲液、无毒相容润滑剂(诸如月桂基硫酸钠和硬脂酸镁)、着色剂、脱模剂、包覆剂、甜味剂、调味剂、芳香剂、防腐剂和抗氧化剂。
[0110]
在一个实施例中,本公开的药物组合物包括一种或多种填充剂、崩解剂和润滑剂。
[0111]
适用于本文所公开的药物组合物的填充剂与药物组合物的其它成分相容,即其基本上不降低药物组合物的溶解度、硬度、化学稳定性、物理稳定性或生物活性。示例性填充剂包含:纤维素、改性纤维素(例如羧甲基纤维素钠、乙基纤维素羟甲基纤维素、羟丙基纤维素)、乙酸纤维素、微晶纤维素、磷酸钙、磷酸氢钙、淀粉(例如玉米淀粉、马铃薯淀粉)、糖(例如,甘露醇、乳糖、蔗糖等)或其任何组合。在一些实施例中,填充剂是微晶纤维素。
[0112]
在一些实施例中,药物组合物包括按药物组合物的重量计,至少25wt%(例如,至少27wt%或至少30wt%)的量的一种或多种填充剂。例如,药物组合物包括按药物组合物的重量计,25wt%至40wt%(例如,25wt%至35wt%或30wt%至33wt%)的填充剂。在另一个实例中,药物组合物包括按药物组合物的重量计,至少25wt%(例如,至少27wt%或至少30wt%)的微晶纤维素,例如avicel ph102或avicel ph101。在另一个实例中,药物组合物包括按药物组合物的重量计,25wt%至40wt%(例如,25wt%至35wt%或30wt%至33wt%)的微晶纤维素。在另一个实例中,药物组合物包括按药物组合物的重量计,约31.7wt%的微晶纤维素。在另一个实例中,药物组合物包括按药物组合物的重量计,约32.6wt%的微晶纤维素。
[0113]
适用于本文所公开的药物组合物的崩解剂可以增强药物组合物的分散并且与药物组合物的其它成分相容,即其基本上不降低药物组合物的化学稳定性、物理稳定性、硬度或生物活性。示例性崩解剂包含交联羧甲基纤维素钠、羟基乙酸淀粉钠、交联聚维酮或其组合。在一些实施例中,崩解剂是交联羧甲基纤维素钠。
[0114]
在一些实施例中,本文所公开的药物组合物包括按药物组合物的重量计,10wt%或更少(例如,8wt%或更少或7wt%或更少)的量的崩解剂。例如,药物组合物包括按药物组合物的重量计,1wt%至10wt%(例如,2wt%至8wt%或3wt%至7wt%)的崩解剂。在另一个实例中,药物组合物包括按药物组合物的重量计,10wt%或更少(例如,8wt%或更少或7wt%或更少)的交联羧甲基纤维素钠。在另一个实例中,药物组合物包括按药物组合物的重量计,1wt%至10wt%(例如,2wt%至8wt%或3wt%至7wt%)的交联羧甲基纤维素钠。在另一个实例中,药物组合物包括按药物组合物的重量计,约5.8wt%的交联羧甲基纤维素钠。在另一个实例中,药物组合物包括按药物组合物的重量计,约6.0wt%的交联羧甲基纤维素钠。
[0115]
在一些实施例中,本文所公开的药物组合物包括润滑剂。润滑剂可以防止混合物组分与表面(例如,混合碗、制粒辊、压缩模具和/或冲头的表面)的粘附。润滑剂还可以减少颗粒内的颗粒间摩擦并改善压缩的药物组合物从制粒机和/或压模机的压缩和喷射。用于本文所公开的药物组合物的合适的润滑剂与药物组合物的其它成分相容,即其基本上不降低药物组合物的溶解度、硬度或生物活性。示例性润滑剂包含硬脂酸镁、硬脂酰富马酸钠、硬脂酸钙、硬脂酸锌、硬脂酸钠、硬脂酸、硬脂酸铝、亮氨酸、山嵛酸甘油酯、氢化植物油或其任何组合。在一些实施例中,润滑剂是硬脂酸镁。
[0116]
在一个实施例中,药物组合物包括按药物组合物的重量计,5wt%或更少(例如,4wt%或更少、3wt%或更少或2wt%或更少)的量的润滑剂。例如,药物组合物包括按药物组合物的重量计,0.10wt%至5wt%(例如,0.5wt%至3wt%或0.75wt%至2wt%)的润滑剂。在另一个实例中,药物组合物包括按药物组合物的重量计,5wt%或更少(例如,4wt%或更少、3wt%或更少或2wt%或更少)的硬脂酸镁。在另一个实例中,药物组合物包括按药物组合物的重量计,0.10wt%至5wt%(例如,0.5wt%至3wt%或0.75wt%至2wt%)的硬脂酸镁。在另一个实例中,药物组合物包括按药物组合物的重量计,约1.0wt%的硬脂酸镁。
[0117]
在一些实施例中,本文所公开的药物组合物是片剂。在一些实施例中,片剂包括膜包衣。在一些实施例中,膜包衣是欧巴代20a100021。在一些实施例中,本文所公开的片剂包括:
[0118][0119]
在一些实施例中,本文所公开的片剂包括:
[0120][0121]
在一些实施例中,本文所公开的片剂包括:
[0122][0123]
在一些实施例中,本文所公开的片剂包括:
[0124][0125][0126]
在一些实施例中,本文所公开的片剂包括:
[0127][0128]
在一些实施例中,本文所公开的片剂包括:
[0129]
[0130]
在一些实施例中,本文所公开的片剂包括:
[0131][0132]
在一些实施例中,本文所公开的片剂包括:
[0133][0134]
在一些实施例中,本文所公开的片剂包括:
[0135]
[0136][0137]
在一些实施例中,本文所公开的片剂包括:
[0138][0139]
在一些实施例中,本文所公开的片剂包括:
[0140]
[0141][0142]
在一些实施例中,本文所公开的片剂包括:
[0143][0144]
一般实验程序
[0145]
除非另有说明,否则通过商业来源获得试剂和起始材料,并且在不纯化的情况下使用。在分别以400mhz和100mhz的1h和
13
c共振频率操作的bruker biospin drx 400mhz ftnmr光谱仪上或在300mhz nmr光谱仪上获得质子和碳nmr光谱。使用broadband observe(bbfo)探针,在20hz样品旋转下,分别以0.1834和0.9083hz/pt数字分辨率获取一维质子和碳谱。使用标准、先前公布的脉冲序列和常规处理参数,在30℃的温度控制下获取所有质子和碳谱。化合物的最终纯度通过反相uplc使用waters制造的acquity uplc beh c
18
柱(50
×
2.1mm,1.7μm颗粒)(pn:186002350)和在3.0分钟内从1-99%流动相b运行的双梯度来测定。流动相a=h2o(0.05%cf3co2h)。流动相b=ch3cn(0.035%cf3co2h)。流速=1.2毫升/分钟,进样体积=1.5μl并且柱温=60℃。通过两个uv迹线(220nm、254nm)的曲线下面积(auc)求平均值来计算最终纯度。使用单四极质谱仪,使用氢离子(h
+
)在检测范围内使用电喷雾电离(esi)获得质量准确度为0.1da并且最小分辨率为1000amu的低分辨率质谱。(2s)-2,4-二甲基-4-硝基-戊酸甲酯的光学纯度是使用手性气体色谱法(gc)分析,在agilent 7890a/msd 5975c仪器上使用restek rt-βdexcst(30m
×
0.25mm
×
0.25um_df)柱以2.0毫升/分钟流速(h2载气)在220℃进样温度和120℃烘箱温度下进行15分钟来确定的。化合物i的纯度通过反相hplc使用poroshell 120ec-c8柱(4.6
×
150mm,2.7μm颗粒)和在40分钟内从30-95%流动相b运行的双梯度来确定。流动相a=5mm乙酸铵ph 4.50并且流动相b=乙腈。流速=1.0毫升/分钟,进样体积=5μl,254nm并且柱温=30℃。
[0146]
化合物i、ii和iii可以通过本领域的任何合适的方法制备。制备化合物i的方法可见于wo 2019/109021和美国专利9,512,079中;制备化合物ii和其药学上可接受的盐的方法公开于wo 2019/161078和pct/us2020/046116中;制备化合物iii和其药学上可接受的盐的方法公开于wo 2011/119984和wo 2011/133751中,所述专利中的所有专利通过引用并入本文。
[0147]
实例1:n-(2-(叔丁基-5-羟基-4-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基-4-氧代-1,4-二氢喹啉-3-甲酰胺(化合物i)的合成
[0148]
(2)(化合物i)的合成的总体方案如下所示,随后是每个中间体的合成的程序。
[0149][0150]
4-(叔丁基)苯-2,6-d2-醇-d(19)的合成的程序
[0151][0152]
向配备有顶置式搅拌器的5l圆底烧瓶中装入4-叔丁基苯酚(14,503.2g)、k2co3(46.3g)、d2o(1949g,1761ml,3.5vol)和meod(409g,503ml,1.0vol)。将混合物在氮气气氛下加热到回流。将反应混合物在回流下老化16小时。将反应混合物冷却至室温并进行取样用于转化(%d并入量)。将反应冷却至5℃并添加35%dc1溶液(90g,71.2ml)。将反应混合物在5℃下老化2小时。将所得浆液过滤并将滤饼用d2o(836g,755ml,1.5vol)洗涤。重复该过程,直到满足目标%d并入量为止(通常需要两个交换)。将湿滤饼在真空烘箱中在40℃下用氮气排放干燥,直到获得恒重为止。4-(叔丁基)苯-2,6-d
2-醇-d(19)的产率为纯度为99.6%并且%d并入量为99.3%的506g的白色固体(98%)。
[0153]
4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯-6-d-醇-d和4-(叔丁基)-2,6-双(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯酚-d(18)的合成的程序
[0154][0155]
将4-(叔丁基)苯-2,6-d
2-醇-d(19)(101.8g,0.66mol,1.0当量)溶解于2l反应器中的ch2ci2(400ml)中。将叔丁醇-£3/4o(43.0g,0.51mol,0.77当量)溶解于250ml烧瓶中的ch2ci2(100ml)中。将叔丁醇-r/溶液在室温下装入2l反应器中。将反应混合物冷却至-5℃。将d2so4(51.1g,0.51mol,0.77当量)通过加料漏斗逐滴装入,同时维持温度范围为-4至-2℃。将反应混合物在-2℃下搅拌3-4小时。在完全转化之后,将反应通过添加水(28ml)淬灭并温热至18-20℃。将底部水层排出并丢弃。将ch2ci2层用饱和nahcch水溶液(大约200ml)处理,将ph调节至6-8。将nacl(饱和)溶液(400ml)装入混合物中。将所得溶液搅拌5分钟,并静置5分钟。将下部ch2ci2层排出到1l烧瓶中。丢弃水层。将ch2ci2溶液浓缩至最小体积并装入正庚烷(200ml)。将溶液浓缩至最小体积并装入庚烷至最终体积为800ml。将0.5n naoh溶液600ml(6vol)装入反应器中并将所得混合物搅拌5分钟,并沉降至少5分钟。将水层排出并丢弃。将1.0n naoh溶液300ml(3vol)装入反应器中并将所得混合物搅拌5分钟,并沉降至少5分钟。将水层排出并丢弃。将1.0n naoh溶液300ml(3vol)装入反应器中并将所得混合物搅拌5分钟,并沉降至少5分钟。将水层排出并丢弃。将剩余的正庚烷溶液浓缩至干燥,得到呈澄清油的期望的产物4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯-6-d-醇-d(18),104.5g,其无需进一步纯化即可进入下一步骤。
[0156]
2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯酚(17)的合成的程序
[0157][0158]
将4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯-6-d-醇-d(18)(100g,0.462mol,1.0当量)溶解于2l反应器中的ch2ci2(800ml,7vol)中并将溶液搅拌。将批料冷却至0
±
3℃。在30分钟内向批料中分批装入n-溴代琥珀酰亚胺(84.4g,0.462mol,1.0当量)。将批料在0
±
2℃下搅拌至少30分钟。然后在2小时的时间段内将批料加热至20
±
2℃并在20
±
2℃下搅拌至少12小时。在完全转化之后,装入饱和nahcch水溶液(500ml,5vol)并将批料搅拌至少10分钟。停止搅动以允许相分离至少5分钟并将ch2ci2层排出,随后将水层去除。将ctlch层装回到容器中。向批料中装入饱和nahcch碳酸氢盐水溶液(500ml,5vol)并将批料搅拌至少10分钟。停止搅动以允许相分离至少5分钟并将ch2ci2层排出,随后将水层去除。将ch2ci2层装回到容器中并用另外的ch2ci2(300ml,3vol)稀释。将批料蒸馏(去除300ml)并通过kf检查以达到干燥。17的所得澄清黄色溶液无需进一步纯化即
可进入下一步骤。
[0159]
2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(16)的合成的程序
[0160][0161]
向清洁反应器中装入4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯-6-d-醇-d(17)(136g,0.462mol,1.0当量)的ch2ci2溶液,随后添加另外的ch2ci2(130ml,1vol),并且搅拌该溶液。向批料中装入4-(二甲基氨基)吡啶(2.8g,0.023mol,0.05当量)和三乙胺(70.1g,0.693mol,1.5当量)。将批料冷却至0
±
3℃。在40分钟内向批料中逐滴装入氯甲酸甲酯(48.0g,0.508mol,1.1当量),同时维持批料温度<5℃。将批料在3
±
2℃下搅拌至少30分钟,并且然后在1小时的时间段内温热至20
±
2℃。在完全转化之后,装入1n hc1(400ml,3vol)。将批料搅拌至少10分钟,并且然后使层分离至少5分钟。将下部有机层排出,随后是水层(第i水层)。将有机层与1n hc1溶液(400ml,3vol)一起装回到反应器中。将批料搅拌至少10分钟,并且然后使层分离至少5分钟。将下部有机层排出。将第一水层与ch2ci2(300ml,2.2vol)一起装入反应器中。将批料搅拌至少10分钟,并且然后使层分离至少5分钟。将下部有机层排出并与第i有机层合并,随后将水层去除。向容器中装入两个有机层的内容物。向反应器中装入水(500ml,3.7vol)。将批料搅拌至少10分钟,并且然后使层分离至少5分钟。将下部有机层排出,随后是水层。将有机层与ch2ci2(400ml,3vol)一起装回到反应器中。将批料蒸馏以去除800ml并通过kf检查以确保干燥。16的所得澄清黄色溶液无需进一步纯化即可进入下一步骤。
[0162]
2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-3-硝基碳酸甲酯(15)的合成的程序
[0163][0164]
向反应器中装入2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(16)并且然后将溶液冷却至0℃。装入硫酸(4.9当量)和硝酸(100%,2.0当量),同时维持温度不超过5℃。将反应在0℃下搅拌2小时直到完全转化为止。然后将反应用水(8.8vol)淬灭并用ch2ci2(1.7vol)稀释。将层分离并将上部水层用ch2ci2(2.8vol)萃取。在
分离层之后,将有机层合并,返回到反应器中并用碳酸氢钠(7.4%w/w,6.8vol)洗涤。在分离层之后,将有机层返回到反应器中并用氯化钠(23%w/w,3.8vol)洗涤。在分离层之后,将有机层返回到反应器中并浓缩至最小体积。装入甲醇(1.2vol),随后浓缩至最小体积。装入甲醇(1.2vol),随后浓缩至最小体积。装入甲醇(1.7vol),并且将浆液加热到回流30分钟并且然后在4小时内缓慢冷却至5℃。将固体产物(15)过滤并将滤饼用冷甲醇(1.0vol)洗涤。将固体2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-3-硝基碳酸甲酯(15)在真空下在40-50℃下干燥,得到类白色固体,纯度为99.9%并且d并入量为99%。
[0165]
5-氨基-4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(7)的合成的程序
[0166][0167]
向反应器中装入5wt%(50-65wt%湿,jm型37)的5%pd/c。装入(4.0vol)甲醇。关闭系统。在2.0巴下用n2
(g
>吹扫。在2.0巴下用h2
(g)
激活。将容器在25℃+/-5℃下用h2
(g
>装到2.0巴。搅拌不少于2小时,同时维持温度为25℃+/-5℃。在2.0巴下用n2
(g)
排气和吹扫。将化合物15(1.0当量)与na2hp04(2.3当量)一起装入反应器中。装入(11.0vol)甲醇。关闭系统。在2.0巴下用n2
(g
>吹扫。在2.0巴下用h2
(g)
激活。将容器在25℃+/-5℃下用h2
(g
>装到2.0巴。搅拌约24小时,同时维持反应温度为25℃+/-5℃。在完全转化之后,通过添加7.7vol的meoh稀释反应混合物。将反应混合物加热至35.0℃+/-5℃。将催化剂和na2hp0
4过滤掉
。将反应器和滤饼用甲醇(4.0vol)洗涤,并过滤,与初始滤液合并。检查pd含量,并且如果需要,进行树脂处理(树脂处理是:装入spm-32树脂(5wt%))。将树脂处理的溶液在35.0℃+/-5℃下搅拌不少于3小时。将树脂过滤掉。
[0168]
将反应器和滤饼用甲醇(2.0vol)洗涤,并过滤,与初始滤液合并。装入norit casp活性碳(3wt%)。在35.0℃+/-5℃下搅拌不少于3小时。将活性碳过滤掉。将反应器和滤饼用甲醇(2.0vol)洗涤,并过滤,与初始滤液合并。在真空下在不超过50℃下蒸馏至8.0vol。装入水(2.0vol),同时维持温度为45℃+/-5℃。在2小时内将所得浆液冷却至0℃+/-5℃。将浆液在0℃+/-5℃下保持并搅拌不少于1小时。将滤饼在0℃+/-5℃下过滤并用2.0体积甲醇/水(8:2)洗涤。将5-氨基-4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(7)在真空下在不超过40℃下干燥,得到白色固体的产物,纯度>99.5%。
[0169]
4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-5-(4-氧代-1,4-二氢喹啉-3-甲酰胺基)苯基碳酸甲酯(8)的合成的程序
[0170][0171]
将化合物7转化成化合物8的程序可以根据化合物5的类似程序来进行。
[0172]
n-(2-(叔丁基)-5-羟基-4-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基)-4-氧代-1,4-二氢喹啉-3-甲散胺基(2)(化全物i)的合成的程序
[0173][0174]
将化合物8转化成化合物2的程序可以根据化合物1的合成的类似程序来进行。
[0175]
实例2:5-氨基-4-(叔丁基-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(7)的合成
[0176]
化合物7的合成的替代总体方案如下所示,随后是每个合成中间体的合成的程序。
[0177][0178]
4-(叔丁基)苯-2,6-d2-醇d(19)的合成的程序
[0179][0180]
向清洁且干燥的500ml反应器中装入4-叔丁基苯酚(14)(24.6g,0.162mmol,1.00当量)、ch2ci2(64ml,2.6vol)和庚烷(64ml,2.6vol),并将该混合物温热至25℃并搅拌,直到所有固体溶解为止。向该溶液中装入氯化氘(35%w/w的氧化氘,25ml,1.0vol),并将该混合物搅拌至少3.5小时。停止搅动并使相分离,并且然后将水层(底部)从反应器中排出。向反应器中装入氯化氘(35%w/w的氧化氘,25ml,1.0vol),并将该混合物搅拌至少3.5小时。停止搅动并使相分离,并且然后将水层(底部)从反应器中排出。向反应器中装入氯化氘(35%w/w的氧化氘,25ml,1.0vol),并将该混合物搅拌至少3.5小时。停止搅动并使相分离,并且然后将水层(底部)从反应器中排出。对所得溶液进行取样并确认相对于起始材料4-叔丁基苯酚至少99%的期望的氘并入产物4-(叔丁基)苯-2,6-d2-醇-d(19)。反应器中的溶液继续进行下一步骤,如下文所描述的。
[0181]
4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯6-d-醇(18)的合成的程序
[0182][0183]
向含有4-(叔丁基)苯-2,6-d2-醇-d(19)的反应混合物的二氯甲烷溶液中装入ch2ci2(125ml,5vol)。使用蒸馏头从反应器中蒸馏出大约125ml的反应溶液并将反应器加热至60℃。向反应器中装入ch2ci2(125ml,5vol)。然后从反应器中蒸馏出大约100ml的反应溶液,并且此时对溶液进行取样以确认水含量(kf)小于300ppm并确定ch2ci2和庚烷含量。在测量批料体积之后,装入ch2ci2(8ml,0.24vol)以将总ch2ci2含量调节至3vol并装入庚烷(68ml,2.8vol)以将庚烷含量调节至4.5vol。向溶液中装入/cvv-乙酸丁酯-dg(30.2g,1.46当量),并将所得溶液冷却至0℃。在至少15分钟内向溶液中装入硫酸-j2(8.12g,0.49当量),并将溶液搅拌2小时,同时维持温度为0-5℃。此后,将温度在两小时内升至20℃并将溶液再搅拌14小时。对溶液进行取样以确认4-叔丁基苯酚(14)或4-(叔丁基)苯-2,6-d2-醇-d(19)以低于3%存在。向反应器中装入ch2ci2(58ml,2.4vol)和庚烷(90ml,3.7vol),并在装入水(125ml,5vol)之前将溶液冷却至0-5℃。将混合物搅拌15分钟,然后停止搅拌并使相分离。从将水相(底部)从反应器中排出之后,装入0.5n naoh水溶液(125ml,5vol)并将温度调节至20℃。将混合物搅拌20分钟,然后停止搅拌并使相分离。对有机相(顶部)进行取样以确认4-叔丁基苯酚(14)或4-(叔丁基)苯-2,6-d2-醇-d(18)以低于0.5%存在。将水相(底部)从反应器中排出。反应器中的溶液继续进行下一步骤,如下文所描述的。
[0184]
2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯酚(17)的合成的
程序
[0185][0186]
在产生4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯-6-d-醇-d(18)的烷基化反应的搅拌的溶液达到0-5℃之后,在至少1小时内装入溴(38.4g,1.45当量),维持温度低于5℃。对溶液进行取样以确认4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯-6-d-醇以低于1%存在。在至少1小时内向溶液中装入焦亚硫酸钠(20%w/w水溶液,147g,0.95当量),维持温度低于10℃。在将温度调节至20℃之后,将混合物再搅拌1小时。停止搅动并使相分离。将水相(底部)从反应器中排出,并将水(125ml,5vol)装入反应器中。将混合物搅拌15分钟,然后停止搅拌并使相分离。将水相(底部)从反应器中排出。反应器中的17的溶液继续进行下一步骤,如下文所描述的。
[0187]
令人惊讶的是,该溴化反应显著提高了硝化反应的选择性。该过程的另一个意想不到的优点是溴化将化合物18和4-(叔丁基)-2,6-双(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯酚的混合物转化为相同的期望的产物(17)。这显著提高了总产率。
[0188]
2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(16)的合成的程序
[0189][0190]
向产生2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯酚(17)的溴化反应的溶液中装入ch2ci2(125ml,5vol)。使用蒸馏头从反应器中蒸馏出大约125ml的反应溶液并将反应器加热至60℃。向反应器中装入ch2ci2(125ml,5vol)。从反应器中蒸馏出大约125ml的反应溶液。向反应器中装入ch2ci2(125ml,5vol)。然后从反应器中蒸馏出大约125ml的反应溶液,并且此时对溶液进行取样以确认水含量(kf)小于300ppm并确定ch2ci2和庚烷含量。在测量批料体积之后,装入cflch以将总ch2ci2含量调节至5.3vol并装入庚烷以将庚烷含量调节至8vol。向溶液中装入三乙胺(31.7g,1.91当量),并将溶液冷却至0-5℃。在至少1小时内向溶液中装入氯甲酸甲酯(24.1g,1.56当量),维持温度低于10℃。将溶液搅拌1小时,并对溶液进行取样以确认2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯酚(17)以低于1%存在。在至少30分钟内向溶液中装入1n盐酸水溶液(125ml,0.76当量),维持温度低于10℃。然后将温度调节至20℃,并且停止搅拌并使相分
离。在将水相(底部)从反应器中排出之后,将水(125ml,5vol)装入反应器中。将混合物搅拌15分钟,然后停止搅拌并使相分离。在将水相(底部)从反应器中排出之后,将水(125ml,5vol)装入反应器中。将混合物搅拌15分钟,然后停止搅拌并使相分离。将水相(底部)从反应器中排出。反应器中的(16)的溶液继续进行下一步骤,如下文所描述的。
[0191]
2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-3-硝基碳酸甲酯(15)的合成的程序
[0192][0193]
向产生2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(16)的保护反应的溶液中装入ch2ci2(125ml,5vol)。使用蒸馏头从反应器中蒸馏出大约125ml的反应溶液并将反应器加热至60℃。向反应器中装入ch2ci2氯化物(125ml,5vol)。从反应器中蒸馏出大约125ml的反应溶液。向反应器中装入ch2ci2(125ml,5vol)。向反应器中装入ch2ci2(125ml,5vol)。从反应器中蒸馏出大约125ml的反应溶液。然后从反应器中蒸馏出大约125ml的反应溶液,并且此时对溶液进行取样以确认水含量(kf)小于300ppm并确定ch2ci2和庚烷含量。在测量批料体积之后,装入ch2ci2以将总ch2ci2含量调节至6vol并装入庚烷以将庚烷含量调节至9vol。在将溶液冷却至0-5℃之后,在至少30分钟内装入硫酸(172g,10.3当量),维持温度低于5℃。在至少30分钟内向混合物中装入硝酸(70%w/w,19.1g,1.31当量),维持温度低于10℃。在将混合物搅拌1小时之后,进行取样并分析以确认2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(16)以低于1%存在。在至少1小时内向混合物中装入水(100ml,4vol),维持温度低于10℃。停止搅拌并使相分离,并将水相(底部)从反应器中排出。在恢复搅拌之后,在至少10分钟内装入碳酸氢钠(8%w/w水溶液,100ml,4vol,0.62当量),维持温度低于10℃。将温度调节至20℃,停止搅拌并使相分离。在将水相(底部)从反应器中排出之后,向反应器中装入水(100ml,4vol)并将混合物搅拌15分钟。停止搅拌,使相分离,并将水相(底部)从反应器中排出。向混合物中装入水(100ml,4vol),并将该混合物搅拌15分钟。停止搅拌,使相分离,并将水相(底部)从反应器中排出。在反应器上标记溶剂水平之后,连接蒸馏头并将温度设定为80℃。向溶液中装入甲醇(570ml,23vol)同时进行蒸馏,通过保持溶剂水平在标记处使添加速率与蒸馏速率相匹配。继续蒸馏,直到批料体积为大约264ml(11vol)为止并且大约1.10kg的蒸馏物已经去除。对混合物进行取样和分析以确认庚烷以低于1%v/v存在。在4小时内将温度调节至0℃。对母液进行取样和分析以确定2-溴-4-(叔丁基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-3-硝基碳酸甲酯(15)的浓度,并将混合物过滤。向反应器中装入甲醇(51.1ml,2vol),并将其搅拌直到温度达到0-5℃为止。该溶液用于洗涤滤饼,并且然后将滤饼通过抽吸干燥至少1小时。然后将固体进行真空干燥,产生呈41.5g的类白色固体的2-溴-4-(叔丁
基)-6-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-3-硝基碳酸甲酯(15)(98.4%纯w/w,纯度校正后的产率为63%)。
[0194]
5-氨基-4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(7)的合成的程序
[0195][0196]
向反应器中装入5wt%(50-65wt%湿,jm型37)的5%pd/c。装入(4.0vol)甲醇。关闭系统。在2.0巴下用n2
(g
>吹扫。在2.0巴下用h2
(g)
激活。将容器在25℃+/-5℃下用h2
(g
>装到2.0巴。搅拌不少于2小时,同时维持温度为25℃+/-5℃。在2.0巴下用n2
(g)
排气和吹扫。将化合物15(1.0当量)与na2hp04(2.3当量)一起装入反应器中。装入(11.0vol)甲醇。关闭系统。在2.0巴下用n2
(g
>吹扫。在2.0巴下用h2
(g)
激活。将容器在25℃+/-5℃下用h2
(g
》装到2.0巴。搅拌约24小时,同时维持反应温度为25℃+/-5℃。在完全转化之后,通过添加7.7vol的meoh稀释反应混合物。将反应混合物加热至35.0℃+/-5℃。将催化剂和na2hp0
4过滤掉
。将反应器和滤饼用甲醇(4.0vol)洗涤,并过滤,与初始滤液合并。检查pd含量,并且如果需要,进行树脂处理(树脂处理是:装入spm-32树脂(5wt%))。将树脂处理的溶液在35.0℃+/-5℃下搅拌不少于3小时。将树脂过滤掉。
[0197]
将反应器和滤饼用甲醇(2.0vol)洗涤,并过滤,与初始滤液合并。装入norit casp活性碳(3wt%)。在35.0℃+/-5℃下搅拌不少于3小时。将活性碳过滤掉。将反应器和滤饼用甲醇(2.0vol)洗涤,并过滤,与初始滤液合并。在真空下在不超过50℃下蒸馏至8.0vol。装入水(2.0vol),同时维持温度为45℃+/-5℃。在2小时内将所得浆液冷却至0℃+/-5℃。将浆液在0℃+/-5℃下保持并搅拌不少于1小时。将滤饼在0℃+/-5℃下过滤并用2.0体积甲醇/水(8:2)洗涤。将5-氨基-4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(7)在真空下在不超过40℃下干燥,得到白色固体的产物,纯度>99.5%。
[0198]
4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-5-(4-氧代-1,4-二氢喹啉-3-甲酰胺基)苯基碳酸甲酯(8)的合成的程序
[0199][0200]
将化合物7转化成化合物8的程序可以根据化合物5的类似程序来进行。
[0201]
n-(2-(叔丁基)-5-羟基-4-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基-4-氧代-1,4-二氢喹啉-3-甲酰胺基(2)(化合物i)的合成的程序
[0202][0203]
将化合物8转化成化合物2的程序可以根据化合物1的合成的类似程序来进行。
[0204]
实例3:5-氨基-4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(7)的合成
[0205]
化合物7的合成的替代方案如下所示,随后是每个合成中间体的合成的程序。
[0206][0207]
5-(叔丁基)-2-羟基苯甲酸(15)的合成的程序
[0208][0209]
将nbuli 1.6m/己烷(3.49g)添加到配备有磁力搅拌棒、热电偶和n2鼓泡器的圆底烧瓶中。将圆底烧瓶冷却至-20℃并开始搅拌。制备2-溴-4-叔丁基苯酚(26)(5.00g)于mtbe
(12.5ml)中的溶液,冷却至-20℃,并逐滴装入圆底烧瓶中,同时维持温度为-20℃+/-5℃。将反应混合物在-20℃+/-5℃下搅拌15分钟,然后温热至23℃。在室温下15分钟之后,通过
′
h nmr(200pl反应混合物稀释到0.75ml d4-meoh中)测量锂化的完全性。当观察到少于1%的2-溴-4-叔丁基苯酚时,认为反应完全。将反应混合物冷却至0℃,添加干冰(固体co2),并将反应在室温下搅拌45分钟。添加水(50.0ml)以将反应淬灭。将混合物转移到分液漏斗中,将相分离,并将有机相丢弃。将水相用1m hc1(15.0ml)酸化至ph为约2,然后用mtbe(25.0ml)萃取三次。将合并的有机萃取物在减压下浓缩,得到呈黄色固体的5-(叔丁基)-2-羟基苯甲酸(25)(2.25g,53.15%产率);3/4nmr(400mhz,d4-meoh):7.86(1h,d,j=2.6hz),7.54(1h,dd,j=8.7,2.6hz),6.85(1h,d,j=2.7hz),1.30(9h,s).
[0210]
5-(叔丁基)-2-羟基苯甲酸甲酯(24)的合成的程序
[0211][0212]
该反应可以根据《生物有机化学与医药化学快报(bioorganic and medicinal chemistry letters)》,2005,第15(21)卷,第4752-4756页中公开的程序进行。
[0213]
2-((叔丁氧基羰基)氧基)-5-(叔丁基)苯甲酸甲酯(23)的合成的程序
[0214][0215]
将碳酸二-e/7-丁基酯(230.55g)和ch2ci2(400ml)装入1l反应器中并搅拌混合物直到固体完全溶解为止。将二甲基氨基吡啶(0.587g)与5-(叔丁基)-2-羟基苯甲酸甲酯(24)(200g)一起装入搅拌的溶液中。将反应混合物在15-30℃下搅拌并在60分钟之后用样品等分试样通过hplc(方法)测量完全性。当5-叔丁基-2-羟基苯甲酸酯(24)的峰面积小于1%时,认为反应完全。通过将饱和氯化铵水溶液(200ml)用水(200ml)稀释,在单独的烧瓶中制备氯化铵的半饱和溶液。将反应混合物用半饱和氯化铵水溶液洗涤两次(每次洗涤200ml)。在每次洗涤期间,将混合物搅拌15分钟并保持15分钟。随后将有机溶液用水洗涤两次(每次洗涤100ml)。在每次洗涤期间,将混合物搅拌15分钟并保持15分钟。将有机溶液转移到1l圆底烧瓶中并在低于35℃和真空下浓缩,得到白色固体(275.51g和99.46%纯度,如通过hplc分析(方法)测量的,93.0%产率的2-((叔丁氧基羰基)氧基)-5-(叔丁基)苯甲酸甲酯(23))。3/4nmr(400mhz,cdcb):8.01(m,1h);7.57(m,1h);7.11(m,1h);3.89(s,3h);1.58(s,9h);1.33(s,9h).
[0216]
4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯酚(22)的合成的程序
[0217][0218]
将thf(176ml)装入500ml夹套反应器中并冷却至5℃。在0-35℃下,向搅拌的溶剂中缓慢装入(甲基-d3)碘化镁(60.5g)于二丁醚(145ml)中的溶液。将所得浆液达到并维持在20-30℃,同时在4-6小内装入2-((叔丁氧基羰基)氧基)-5-(叔丁基(苯甲酸酯(23)(22g)于thf(44ml)中的溶液。将反应混合物在20-30℃下搅拌并在60分钟之后用样品等分试样通过hplc测量完全性。当2-((叔丁氧基羰基)氧基)-5-(叔丁基)苯甲酸酯(23)的峰面积小于1%时,认为反应完全。向第二反应器中装入6n盐酸水溶液(110ml)并将搅拌的溶液冷却至0-10℃。将反应浆液缓慢转移到0-35℃的酸溶液中。将相搅拌15分钟并在分离之前保持15分钟。将水相用二丁醚(132ml)萃取。在萃取期间,将相搅拌15分钟并在分离之前保持15分钟。将合并的有机相依次用水(2
×
77ml)、5%硫代硫酸钠水溶液(77ml)和水(77ml)洗涤。在每次洗涤期间,将混合物搅拌15分钟并保持15分钟。将有机溶液转移到圆底烧瓶中并在低于80℃和真空下浓缩,得到呈粗制油状物的4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯酚(22)(5.94g和83.8%纯度,如通过hplc分析测量的,通过lc/ms分析具有99.3%的d9同位素纯度,84.9%产率的甲基4-(叔丁基)-2-(2-(甲基-i3/4)丙-2-基-1,1,1,3,3,3-^苯酚(23))。3/4nmr(400mhz,cd3od):7.22(m,1h);7.00(m,1h);6.65(m,1h);1.26(s,9h).
[0219]
(23)的格氏反应导致一些氘并入(22)中。为了实现h/d交换,将混合物进行一系列hc1洗涤:
[0220][0221]
h/d交换程序
[0222]
将化合物22的氘化类似物(1.00当量)装入反应器中。装入dcm(5vol)。将夹套设置为20℃。搅拌至固体溶解。装入35%盐酸(5vol)。搅拌混合层不少于6小时。停止搅拌并使层沉降至少30分钟。将底层(有机物)从反应器中排出。将水层从反应器中排出。将有机部分装回到反应器中。重复hcl洗涤序列两次。装入预混合水(2.5vol)和饱和nacl水溶液(2.5vol)。搅拌混合层30分钟。停止搅拌并使层沉降至少30分钟。将底层(有机物)从反应器中排出。将水溶液从反应器中排出。将有机部分装回到反应器中。装入水(5vol)。搅拌混合层30分钟。停止搅拌并使层沉降至少30分钟。将底层(有机物)从反应器中排出。将水溶液从
反应器中排出。将有机部分装回到反应器中。将溶剂在减压下蒸馏至最小体积(使用水浴温度为35℃的旋转蒸发器)。装入dcm(5vol)。将溶剂在减压下蒸馏至最小体积(使用水浴温度为35℃的旋转蒸发器)。装入dcm(5vol)。对溶液进行取样并通过kf测量水含量。重复操作直到水含量低于300ppm为止。注意:该溶液直接用于下一步反应,因此dcm的最终量应该是化合物22的烷氧基甲酰化反应所需的量。
[0223]
4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(21)的合成的程序
[0224][0225]
将化合物22转化成化合物21的程序可以根据化合物12的类似程序来进行。
[0226]
4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-5-硝基碳酸甲酯(20)的合成的程序
[0227][0228]
将化合物21转化成化合物20的程序可以根据化合物11a的类似程序来进行。
[0229]
5-氨基-4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基碳酸甲酯(7)的合成的程序
[0230][0231]
将化合物20转化成化合物7的程序可以根据化合物4的类似程序来进行。
[0232]
4-(叔丁基)-2-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)-5-(4-氧代-1,4-二氢喹啉-3-甲酰胺基)苯基碳酸甲酯(8)的合成的程序
[0233][0234]
将化合物7转化成化合物8的程序可以根据化合物5的类似程序来进行。
[0235]
n-(2-(叔丁基)-5-羟基-4-(2-(甲基-d3)丙-2-基-1,1,1,3,3,3-d6)苯基)-4-氧代-1,4-二氢喹啉-3-甲酰胺基(2)(化合物i)的合成的程序
[0236][0237]
将(8)转化成(2)的程序可以根据化合物i的合成的类似程序来进行。
[0238]
实例4:(14s)-8-[3-(2-{二螺[2.0.2.1]庚-7-基}乙氧基)-1h-吡唑-1-基]-12,12-二甲基-2λ6-硫杂-3,9,11,18,23-五氮杂四环[17.3.1.111,14.05,10]二十四碳-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮(化合物ii)的合成
[0239]
除非另有说明,否则通过商业来源获得试剂和起始材料,并且在不纯化的情况下使用。
[0240]
在分别以400mhz和100mhz的1h和
13
c共振频率操作的bruker biospin drx 400mhz ftnmr光谱仪上或在300mhz nmr光谱仪上获得质子和碳nmr光谱。使用broadband observe(bbfo)探针,在20hz样品旋转下,分别以0.1834和0.9083hz/pt数字分辨率获取一维质子和碳谱。使用标准、先前公布的脉冲序列和常规处理参数,在30℃的温度控制下获取所有质子和碳谱。
[0241]
部分a:2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酸的合成
[0242][0243]
步骤1:7-(溴甲基)二螺[2.0.2.1]庚烷
[0244][0245]
使1000ml、3颈圆底烧瓶配备有机械搅拌器、冷却浴、加料漏斗、j-kem温度探针和氮气入口/出口。在氮气气氛下,向容器中装入三苯基膦(102.7ml,443.2mmol)和二氯甲烷(1l),得到澄清无色溶液。开始搅拌,并向冷却浴中装入丙酮。将干冰分批添加到冷却浴中,直到获得-15℃的锅温。向加料漏斗中装入溴(22.82ml,443.0mmol)于二氯甲烷(220ml,10ml/g)中的溶液,随后在1小时内逐滴添加该溶液。在添加期间分批向冷却浴中添加干冰以维持锅釜温度在-15℃。在溴添加完成后,继续在-15℃下将浅黄色悬浮液搅拌15分钟,此时将悬浮液冷却至-30℃。向加料漏斗中装入二螺[2.0.2.1]庚-7-基甲醇(50g,402.6mmol)、吡啶(35.82ml,442.9mmol)和二氯甲烷(250ml,5ml/g)的溶液。接着历经1.5小时逐滴添加澄清浅黄色溶液,维持锅釜温度在-30℃。使所得澄清浅黄色反应混合物逐渐升温至-5℃的锅釜温度并且然后继续在-5℃下搅拌1小时。然后将反应混合物倒入己烷(2000ml)中,导致沉淀的形成。将悬浮液在室温下搅拌30分钟,并且然后通过具有20mm硅藻土层的玻璃料布氏漏斗过滤。将澄清滤液减压浓缩(水浴温度为20℃),得到含有一些沉淀的黄色油状物。将油状物用一些己烷稀释,在室温下静置15分钟,然并且后通过具有20mm硅藻土层的玻璃料布氏漏斗过滤。将澄清滤液减压浓缩(水浴温度为20℃),得到呈澄清黄色油状物的7-(溴甲基)二螺[2.0.2.1]庚烷(70g,93%)。1h nmr(400mhz,氯仿-d)δ3.49(d,j=7.5hz,2h),1.90(t,j=7.5hz,1h),1.06-0.84(m,4h),0.71(ddd,j=9.1,5.1,4.0hz,2h),0.54(dddd,j=8.6,4.8,3.8,1.0hz,2h)。
[0246]
步骤2:2-二螺[2.0.2.1]庚-7-基乙腈
[0247][0248]
使1000ml、3颈圆底烧瓶配备有机械搅拌器、用作二级防护的冷却浴、j-kem温度探针和氮气入口/出口。在氮气气氛下,向容器中装入7-(溴甲基)二螺[2.0.2.1]庚烷(35g,187.1mmol)和二甲基亚砜(245ml),得到澄清琥珀色溶液。开始搅拌,并记录锅温为19℃。然后向容器装入一次性添加的固体氰化钠(11.46g,233.8mmol),得到深色溶液并在15分钟内逐渐放热至49℃。几分钟后,锅釜温度开始降低,并且将混合物继续在室温下搅拌过夜(约15小时)。将深色反应混合物用冰冷饱和碳酸钠溶液(500ml)淬灭,并且然后转移到分液漏
斗中并用乙醚(500ml)分配。移除有机物,并用乙醚(2x 250ml)萃取残余水。将合并的有机物用水(500ml)洗涤,经硫酸钠(200g)干燥,并且然后通过玻璃料布氏漏斗过滤。将澄清琥珀色滤液减压浓缩(水浴温度20℃),得到呈澄清琥珀色油状物的2-二螺[2.0.2.1]庚-7-基乙腈(21g,84%)。1h nmr(400mhz,氯仿-d)δ2.42(d,j=6.6hz,2h),1.69(t,j=6.6hz,1h),1.02-0.88(m,4h),0.79-0.70(m,2h),0.66-0.55(m,2h)。
[0249]
步骤3:2-二螺[2.0.2.1]庚-7-基乙酸
[0250][0251]
向2-二螺[2.0.2.1]庚-7-基乙腈(2.1g,14.19mmol)于etoh(32ml)中的溶液中添加氢氧化钠(5.12g,128.0mmol),随后添加水(13ml),并将所得溶液搅拌并加热至70℃过夜。然后将混合物冷却至室温,用水稀释并用乙醚萃取。通过添加6n盐酸(产生浑浊沉淀)将水相调节至ph=1,并用乙醚(3x)萃取。将有机相干燥(硫酸镁)、过滤并浓缩,得到呈橙色固体状的2-二螺[2.0.2.1]庚-7-基乙酸(2.19g,99%产率,98%纯度),其不经进一步纯化即用于下一步。1h nmr(400mhz,氯仿-d)δ2.44(d,j=6.9hz,2h),1.67(t,j=6.9hz,1h),0.91(ddd,j=9.0,5.2,3.9hz,2h),0.81(dddd,j=8.9,5.2,3.9,0.5hz,2h),0.69(ddd,j=8.9,5.2,3.9hz,2h),0.56-0.44(m,2h)。
[0252]
步骤4:2-二螺[2.0.2.1]庚-7-基乙醇
[0253][0254]
在15分钟内向在冰/水浴中冷却的溶解于四氢呋喃(33.71ml)中的氢化铝锂(827.4mg,902.3μl,21.80mmol)中逐滴添加含2-二螺[2.0.2.1]庚-7-基乙酸(2.552g,16.77mmol)的四氢呋喃(7.470ml),保持反应温度《20℃。使混合物搅拌总共18小时,逐渐升温到环境温度。将混合物用冰/水浴冷却,并且通过依次缓慢添加水(838.4mg,838.4μl,46.54mmol),随后添加氢氧化钠(1.006ml,5m,5.031mmol),然后添加水(2.493g,2.493ml,138.4mmol)淬灭,得到经硅藻土过滤的白色粒状浆液。用乙醚洗涤过滤后的固体。将滤液在约300mbar和30℃水浴下真空浓缩。将残余物用乙醚稀释,干燥(硫酸镁),过滤并在约300mbar和30℃水浴下真空浓缩,随后真空浓缩约30秒,得到2-二螺[2.0.2.1]庚-7-基乙醇(2.318g,100%),其不经进一步纯化即直接用于后续步骤。1h nmr(400mhz,氯仿-d)δ3.64(s,2h),1.68(d,j=6.7hz,2h),1.39(s,1h),1.31(s,1h),0.82(d,j=14.0hz,4h),0.65(s,2h),0.50(d,j=3.6hz,2h)。
[0255]
步骤5:3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-甲酸叔丁酯
[0256][0257]
向5-氧代-1h-吡唑-2-甲酸叔丁酯(2.942g,15.97mmol)和2-二螺[2.0.2.1]庚-7-基乙醇(2.318g,16.77mmol)于四氢呋喃(36.78ml)中的溶液中添加三苯基膦(4.399g,16.77mmol)。在10分钟内向混合物中缓慢地逐滴添加偶氮二甲酸二异丙酯(3.391g,3.302ml,16.77mmol)(注意到轻度放热)。将反应混合物在室温下搅拌30分钟,然后在50℃
下搅拌30分钟。真空依次四氢呋喃。向粗残余物中添加甲苯(23.54ml),并将混合物搅拌过夜,因为沉淀逐渐结晶。用硅藻土浆化,然后将沉淀过滤掉,并用甲苯(8.705ml)洗涤,并且再用甲苯(8.705ml)洗涤。将滤液真空浓缩。通过硅胶色谱法,使用从100%己烷到100%乙酸乙酯的窄梯度来纯化粗产物,得到3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-甲酸叔丁酯(3.449g,71%)。esi-ms m/z计算值304.17868,实验值305.1(m+1)
+
;保留时间:0.82分钟(lc方法a)。
[0258]
步骤6:3-(2-二螺[2.0.2.1]庚-7-基乙氧基)-1h-吡唑
[0259][0260]
将3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-甲酸叔丁酯(5.304g,17.43mmol)溶解于含有三氟乙酸(29.81g,20.14ml,261.4mmol)的二氯甲烷(53.04ml)中,并将反应物在室温下搅拌120分钟。将反应物蒸发,并且将所得油状物在乙酸乙酯和饱和碳酸氢钠溶液之间分配,并分离各层。将水性部分用乙酸乙酯萃取两次,然后将有机物合并,用盐水洗涤,经硫酸钠干燥,过滤并蒸发,得到油状物3-(2-二螺[2.0.2.1]庚-7-基乙氧基)-1h-吡唑(3.56g,100%)。esi-ms m/z计算值204.12627,实验值205.1(m+1)
+
;保留时间:0.59分钟(lc方法a)。
[0261]
步骤7:2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酸叔丁酯
[0262][0263]
将2,6-二氯吡啶-3-甲酸叔丁酯(4.322g,17.42mmol)、3-(2-二螺[2.0.2.1]庚-7-基乙氧基)-1h-吡唑(3.559g,17.42mmol)和碳酸钾(2.891g,20.92mmol)合并于无水二甲基亚砜(71.18ml)中。添加1,4-二氮杂双环[2.2.2]辛烷(391.1mg,3.487mmol),并将混合物在室温下在氮气下搅拌16小时。将反应混合物用水(136.9ml)稀释并搅拌15分钟。将所得白色固体过滤并用水洗涤。将固体溶解于二氯甲烷中并经硫酸镁干燥。将混合物过滤并蒸发,得到呈白色固体状的2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酸叔丁酯(5.69g,79%)。1h nmr(400mhz,氯仿-d)δ8.35(d,j=2.9hz,1h),8.18(d,j=8.4hz,1h),7.69(d,j=8.4hz,1h),5.94(d,j=2.9hz,1h),4.25(s,2h),1.90(d,j=6.8hz,2h),1.62(s,9h),1.49(t,j=6.6hz,1h),0.85(d,j=1.5hz,4h),0.65(d,j=1.5hz,2h),0.52(d,j=1.1hz,2h)。esi-ms m/z计算值415.16626,实验值360.0(m-tbu)+;保留时间:2.09分钟(lc方法b)。
[0264]
步骤8:2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酸
[0265][0266]
将2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酸叔丁酯(5.85g,14.07mmol)溶解于含有三氟乙酸(16.26ml,211.1mmol)的二氯甲烷(58.5ml)中,并将反应物在室温下搅拌16小时。将反应物蒸发,并向所得固体中添加醚,并且然后减压移除醚。再重复两次从醚中蒸发,得到白色固体2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酸(5.06g,100%)。1h nmr(400mhz,氯仿-d)δ8.41(d,j=8.5hz,1h),8.37(d,j=2.9hz,1h),7.75(d,j=8.5hz,1h),5.97(d,j=2.9hz,1h),4.27(s,2h),1.91(d,j=6.7hz,2h),1.50(s,1h),0.85(d,j=1.5hz,4h),0.71-0.62(m,2h),0.52(d,j=1.1hz,2h).esi-ms m/z calc.359.10367,实验值360.2(m+1)+;保留时间:2.16分钟(lc方法b)。
[0267]
部分b(4s)-2,2-二甲基-4-[3-[(6-氨磺酰基-2-吡啶基)氨基]丙基]吡咯烷-1-甲酸叔丁酯的合成
[0268][0269]
步骤1:(e)-(2-氧代四氢吡喃-3-亚基)甲醇盐(钠盐)
[0270][0271]
使5l、3颈圆底烧瓶配备有机械搅拌器、加热套、加料漏斗、j-kem温度探针/控制器和氮气入口/出口。在氮气气氛下,向容器中装入氢化钠(59.91g,60%w/w,1.498mol),随后装入庚烷(1.5l),得到灰色悬浮液。开始搅拌,并记录锅温为19℃。然后向容器中装入通过注射器添加的乙醇(3.451g,74.91mmol),此导致气体逸出。向加料漏斗中装入四氢吡喃-2-酮(150g,1.498mol)和甲酸乙酯(111g,1.50mol)的澄清浅黄色溶液。历经1小时逐滴添加该溶液,此导致气体逸出并逐渐放热至45℃。然后将所得浓稠白色悬浮液加热至65℃持续2小时,并且然后使其冷却至室温。将混合物在室温下继续搅拌过夜(约10小时)。将反应混合物在氮气流下通过玻璃料布氏漏斗(中等孔隙度)真空过滤。将滤饼用庚烷(2x 250ml)置换洗涤并抽吸几分钟。将略带庚烷的湿滤饼转移到玻璃托盘中,并且在真空烘箱中于45℃下干燥15小时,得到作为所需产物(e)-(2v氧代四氢吡喃-3-亚基)甲醇盐(钠盐)的白色固体
(205g,1.36mol,91%产率)。
[0272]
步骤2:3-亚甲其四氢吡喃-2-酮
[0273][0274]
使5l、3颈圆底烧瓶配备有机械搅拌器、加热套、加料漏斗、j-kem温度探针/控制器和氮气入口/出口。在氮气气氛下,向容器中装入(e)-(2-氧代四氢吡喃-3-亚基)甲醇盐(钠盐)(205g,1.366mol)(205g,1.366mol)和四氢呋喃(1640ml),得到白色悬浮液。开始搅拌,并记录锅温为19℃。然后向容器中装入一次性添加的固体多聚甲醛(136.6g,4.549mol)。将所得悬浮液加热至63℃,并将条件保持15小时。在加热后,反应混合物变成略带明胶状。将白色明胶状混合物减压浓缩以去除大部分四氢呋喃。将剩余残留物在分液漏斗中用乙酸乙酯(1000ml)、饱和氯化钠(500ml)和饱和碳酸钠(500ml)分配。移除有机物,并用乙酸乙酯(5x 300ml)萃取残余水。将合并的有机物经硫酸钠(500g)干燥,并且然后通过带有20mm硅藻土层的玻璃料布氏漏斗真空过滤。将滤饼用乙酸乙酯(250ml)置换洗涤。将澄清滤液减压浓缩,得到作为所需粗产物的澄清浅黄色油状物(135g)。材料通过硅胶柱快速色谱法(液体装载),用在1小时内100%己烷至60%乙酸乙酯/己烷的梯度洗脱进行纯化,收集到450ml级分。通过tlc分析在硅胶上用3:1己烷/乙酸乙酯洗脱来检测产物,并且在uv下可视化。将产物级分合并并且减压浓缩,得到作为所需产物3-亚甲基四氢吡喃-2-酮的澄清无色油状物(132g,1.18mol,72%产率,由nmr测定含有16wt%残留乙酸乙酯)。1h nmr(400mhz,二甲基亚砜-d6)δ6.18(q,j=1.9hz,1h),5.60(q,j=1.9hz,1h),4.40-4.26(m,2h),2.61(ddt,j=7.0,6.3,2.0hz,2h),1.90-1.75(m,2h)。
[0275]
步骤3:3-(2-甲基-2-硝基-丙基)四氢吡喃-2-酮
[0276][0277]
使5000ml、3颈圆底烧瓶配备有机械搅拌器、用作二级防护的冷却浴、j-kem温度探针、加料漏斗和氮气入口/出口。在氮气气氛下,向容器中装入2-硝基丙烷(104.9g,1.177mol)。开始搅拌,并记录锅温为19℃。然后向容器中装入整份添加的纯1,8-二氮杂双环[5.4.0]十一碳-7-烯(22.41g,147.2mmol),得到澄清浅黄色溶液。未观察到放热。向加料漏斗中装入3-亚甲基四氢吡喃-2-酮(110g,981.0mmol)于乙腈(1100ml)中的溶液,其历经1小时逐滴添加,产生澄清浅黄色溶液并逐渐放热至24℃。将反应混合物继续在室温下搅拌3.5小时并且然后减压浓缩。将剩余的残留物溶解于二氯甲烷(1000ml)中,并用500ml的1摩尔柠檬酸溶液/饱和氯化钠溶液的3:2混合物分配。所得有机相为澄清浅蓝色溶液,并且水相为略浑浊的极淡蓝色溶液。移除有机物,并用二氯甲烷(300ml)萃取残余水溶液。将合并的有机物用饱和氯化钠溶液(300ml)洗涤,经硫酸钠(250g)干燥,并且然后通过玻璃料布氏漏斗过滤。将滤液减压浓缩至约200ml的体积。将澄清浅蓝色二氯甲烷溶液用甲基叔丁基醚(1500ml)稀释,并且将浑浊溶液减压浓缩至约200ml的体积,由此得到悬浮液。将混合物再次用甲基叔丁基醚(1500ml)稀释,并减压浓缩至约250ml的体积。使所得悬浮液在室温下静置过夜(约12小时)。通过在玻璃料布氏漏斗中真空过滤收集固体,并将滤饼用冷的甲基叔丁基醚(2x 150ml)置换洗涤,并且然后抽吸30分钟。将材料在真空烘箱中于45℃下进一步
干燥5小时,得到(160g,0.795mol,81%产率)作为所需产物3-(2-甲基-2-硝基-丙基)四氢吡喃-2-酮的白色固体。1h nmr(400mhz,二甲基亚砜-d6)δ4.34(ddd,j=11.1,9.3,4.3hz,1h),4.20(dt,j=11.1,5.1hz,1h),2.75-2.62(m,1h),2.56(dd,j=14.9,5.2hz,1h),2.01-1.89(m,2h),1.89-1.67(m,2h),1.55(d,j=6.0hz,6h),1.44(dddd,j=12.8,11.5,8.1,6.6hz,1h)。
[0278]
步骤4:3-(3-羟丙基)-5,5-二甲基-吡咯烷-2-酮
[0279][0280]
使1000ml、3颈圆底烧瓶配备有特氟隆搅拌棒、加热套、j-kem温度探针/控制器和橡胶隔片。向容器中装入3-(2-甲基-2-硝基-丙基)四氢吡喃-2-酮(25g,124.2mmol)和乙醇(375ml),得到白色悬浮液。开始搅拌,并将悬浮液加热至40℃持续10分钟,得到澄清无色溶液。然后使容器配备有气体分散管,并将溶液用氮气脱气15分钟。然后向容器中装入雷尼镍(raney nickel)(8.019g,50%w/w,68.31mmol),并且然后使容器配备有隔片。将容器抽真空并置于氢气气氛下。重复该过程三个循环。然后将容器置于1个大气压的氢气下并且将反应混合物逐渐加热至60℃。将反应物继续在60℃下搅拌24小时。在冷却到室温之后,使容器配备有气体分散管,并将反应混合物用氮气脱气15分钟。将混合物通过带有20mm硅藻土层的玻璃料布氏漏斗进行真空过滤。将滤饼用乙醇(2x 100ml)置换洗涤,并抽吸直到略微乙醇润湿,然后用水润湿,并在水下丢弃使用过的雷尼镍催化剂。将澄清浅琥珀色滤液减压浓缩至澄清粘性浅琥珀色油状物。将油状物用甲基叔丁基醚(1500ml)稀释,并且将浑浊溶液减压浓缩至约150ml的体积,由此得到悬浮液。将混合物再次用甲基叔丁基醚(1500ml)稀释,并减压浓缩至约150ml的体积。使所得悬浮液在室温下静置过夜(约12小时)。通过在玻璃料布氏漏斗中真空过滤收集固体,并将滤饼用冷的甲基叔丁基醚(2x 50ml)置换洗涤,并且然后抽吸30分钟。将材料在真空烘箱中于45℃下进一步干燥3小时,得到作为所需产物3-(3-羟丙基)-5,5-二甲基-吡咯烷-2-酮的白色固体(19g,0.111mol,89%产率)。1h nmr(400mhz,二甲基亚砜-d6)δ7.63(s,1h),3.38(t,j=6.5hz,2h),2.37(tdd,j=9.8,8.5,4.4hz,1h),2.02(dd,j=12.3,8.6hz,1h),1.72(tdd,j=9.6,7.5,4.4hz,1h),1.52-1.32(m,3h),1.28-1.03(m,7h)。
[0281]
步骤5:3-(5,5-二甲基吡咯烷-3-基)丙-1-醇
[0282][0283]
使5l、3颈圆底烧瓶配备有机械搅拌器、加热套、加料漏斗、j-kem温度探针/控制器和氮气入口/出口。在氮气气氛下,向容器中装入氢化铝锂粒料(19.39g,510.9mmol)。然后向容器中装入四氢呋喃(500ml,20ml/g)。开始搅拌,并记录锅温为20℃。将混合物在室温下搅拌0.5小时以允许团粒溶解。所得灰色悬浮液的锅釜温度记录为24℃。向加料漏斗中装入3-(3-羟丙基)-5,5-二甲基-吡咯烷-2-酮(25g,146.0mmol)于四氢呋喃(500ml)中的溶液,并且历经90分钟逐滴添加澄清浅黄色溶液。需要轻微加热以实现均质性。在添加完成后,所得淡灰色悬浮液的锅釜温度记录为24℃。然后将混合物加热至65℃的锅釜温度,并且将该条件维持72小时。此时,对反应混合物的分析表明,仍然保留一些残余的起始材料,并且产
物形成没有变化。随后,反应在此时停止。移除加热套,并且使容器配备有冷却浴。将悬浮液用碎冰/水冷却浴冷却至0℃,并且然后通过极缓慢逐滴添加水(19.93ml),随后添加15wt%的氢氧化钠溶液(19.93ml),最后用水(59.79ml)淬灭。所得白色悬浮液的锅釜温度记录为5℃。将冷却浴移除,并再次向容器装配加热包。将悬浮液温热至60℃,并将条件保持30分钟。将热悬浮液通过带有20mm硅藻土层的玻璃料布氏漏斗进行真空过滤。然后将滤饼用60℃四氢呋喃(2x 250ml)置换洗涤,并且然后抽吸30分钟。将澄清滤液减压浓缩,得到(23.5g,0.149mol,99%产率)的3-(5,5-二甲基吡咯烷-3-基)丙-1-醇的澄清浅黄色粘性油状物作为所需产物。1h nmr(400mhz,二甲基亚砜-d6)δ3.37(dt,j=8.3,6.4hz,3h),2.95(dd,j=10.6,7.6hz,1h),2.40(dd,j=10.7,7.7hz,1h),2.04(dt,j=16.1,8.1hz,1h),1.69(dd,j=12.2,8.2hz,1h),1.50-1.24(m,5h),1.11-0.94(m,7h)。
[0284]
步骤6:4-(3-羟丙基)-2,2-二甲基-吡咯烷-1-甲酸叔丁酯
[0285][0286]
使1l、3颈圆底烧瓶配备有机械搅拌器、冷却浴、加料漏斗、j-kem温度探针和氮气入口/出口。在氮气气氛下,向容器中装入3-(5,5-二甲基吡咯烷-3-基)丙-1-醇(15g,95.39mmol)和二氯甲烷(225ml,15ml/g),得到澄清浅黄色溶液。开始搅拌,并记录锅温为19℃。向冷却浴中装入碎冰/水,并且将锅温降低至0℃。向加料漏斗中装入三乙胺(12.55g,124.0mmol),其随后历经5分钟逐滴纯净地添加。未观察到放热。然后向加料漏斗中装入溶解于二氯甲烷(225ml)中的二碳酸二叔丁酯(22.89g,104.9mmol)。然后在30分钟内逐滴添加澄清浅黄色溶液,从而导致缓慢气体逸出。未观察到放热。移除冷却浴,并且使所得的澄清浅黄色溶液温热至室温,并在室温下继续搅拌3小时。将反应混合物转移到分液漏斗中并用水(75ml)分配。将有机物移除并用饱和氯化钠溶液(75ml)洗涤,经硫酸钠(150g)干燥,并且然后通过玻璃料布氏漏斗过滤。将滤液减压浓缩,得到(30g)作为所需粗产物的澄清浅黄色油状物。该材料通过硅胶柱快速色谱法(液体负载用二氯甲烷进行),在60分钟内用100%二氯甲烷至10%甲醇/二氯甲烷的梯度洗脱进行纯化,从而收集50ml级分。将期望的产物级分合并,并在减压下浓缩,得到呈澄清淡黄色粘性油状物的4-(3-羟丙基)-2,2-二甲基-吡咯烷-1-甲酸叔丁酯(22g,0.0855mol,90%产率)。1h nmr(400mhz,dmso-d6)δ4.38(td,j=5.2,1.4hz,1h),3.54(dt,j=10.3,6.7hz,1h),3.38(td,j=6.6,3.5hz,2h),2.76(q,j=10.3hz,1h),2.07(td,j=11.6,5.7hz,1h),1.87(ddd,j=16.7,12.1,6.0hz,1h),1.37(dd,j=14.2,10.4hz,17h),1.24(s,3h)。
[0287]
步骤7:2,2-二甲基-4-(3-甲基磺酰基氧基丙基)吡咯烷-1-甲酸叔丁酯
[0288][0289]
将4-(3-羟丙基)-2,2-二甲基-吡咯烷-1-甲酸叔丁酯(50.5g,196.22mmol)和三乙胺(39.711g,54.698ml,392.44mmol)溶解于二氯甲烷(500ml)中,并将所得溶液在冰水浴中冷却30分钟。在30分钟内逐滴添加甲磺酰氯(24.725g,16.706ml,215.84mmol),然后移除冰浴,并将混合物在室温下搅拌一小时。然后用饱和碳酸氢钠溶液(200ml)淬灭反应物。分离各相,并用饱和碳酸氢钠(200ml)和水(2x 100ml)萃取有机相。将水相丢弃,并将有机相经硫酸钠干燥,过滤并真空浓缩,得到呈浅黄色油状物的2,2-二甲基-4-(3-甲基磺酰基氧基
丙基)吡咯烷-1-甲酸叔丁酯(64.2g,93%)。esi-ms m/z计算值335.1766,实验值336.4(m+1)
+
;保留时间:5.54分钟(lc方法q)。
[0290]
步骤8:4-(3-氨基丙基)-2,2-二甲基-吡咯烷-1-甲酸叔丁酯
[0291][0292]
将2,2-二甲基-4-(3-甲基磺酰基氧基丙基)吡咯烷-1-甲酸叔丁酯(64.2g,191.38mmol)溶解在二噁烷(650ml)中,并且然后添加氢氧化铵(650ml),并将所得混合物加热至45℃,持续18小时。在18小时后,将反应物冷却至室温。将溶液用1m氢氧化钠(200ml)稀释,并且然后用乙醚(3x 650ml)萃取。丢弃水相,并且用水(2x 200ml)萃取合并的有机相。将水相丢弃,并将有机相经硫酸钠干燥,过滤并真空浓缩,得到呈浅黄色油状物的4-(3-氨基丙基)-2,2-二甲基-吡咯烷-1-甲酸叔丁酯(48.9g,95%)。esi-ms m/z计算值256.2151,实验值257.3(m+1)
+
;保留时间:3.70分钟(lc方法q)。
[0293]
步骤9:2,2-二甲基-4-[3-[(6-氨磺酰基-2-吡啶基)氨基]丙基]吡咯烷-1-甲酸叔丁酯
[0294][0295]
向含4-(3-氨基丙基)-2,2-二甲基-吡咯烷-1-甲酸叔丁酯(8.91g,34.8mmol)和6-氟吡啶-2-磺酰胺(6.13g,34.8mmol)的二甲基亚砜(75ml)中添加碳酸钾(4.91g,35.5mmol),并且将混合物在100℃下搅拌12小时,并且然后冷却至环境温度并再搅拌4小时(总共16小时)。将反应混合物缓慢倒入含盐酸(35ml,1m,35.00mmol)的水(200ml)中(有些起泡沫),并用乙酸乙酯(250ml)稀释。分离有机相并用100ml盐水洗涤。将有机相经硫酸镁干燥,经硅藻土过滤并真空浓缩,得到深黄色油状物。粗产物通过硅胶色谱法,用0%-100%乙酸乙酯/己烷洗脱来进行纯化。收集纯(9.0g)和不纯(3g)级分。通过硅胶色谱法,用0%-100%乙酸乙酯/己烷洗脱来纯化不纯级分,总共得到2,2-二甲基-4-[3-[(6-亚氨基甲酰基-2-吡啶基)氨基]丙基]吡咯烷-1-甲酸叔丁酯(10.0g,69%)。1h nmr(400mhz,二甲基亚砜-d6)δ7.52(dd,j=8.5,7.2hz,1h),7.07(s,2h),6.95(dd,j=7.2,0.7hz,2h),6.61(d,j=8.5hz,1h),3.55(q,j=9.1hz,1h),3.32-3.24(m,2h),2.79(q,j=10.0hz,1h),2.13(d,j=16.1hz,1h),1.96-1.82(m,1h),1.51(dt,j=18.0,9.3hz,2h),1.37(dd,j=12.9,10.6hz,15h),1.24(s,3h).esi-ms m/z calc.412.21442,实验值413.1(m+1)
+
;保留时间:2.34分钟(lc方法d)。
[0296]
步骤10:(4s)-2,2-二甲基-4-[3-[(6-氨磺酰基-2-吡啶基)氨基]丙基]吡咯烷-1-甲酸叔丁酯
[0297][0298]
通过sfc色谱法,使用chiralpak ig(250x 21.2mm柱,5μm粒度)和40%甲醇/60%
二氧化碳流动相,以70毫升/分钟在11.0分钟内(进样体积=500μl的32mg/ml于甲醇中的溶液)对外消旋2,2-二甲基-4-[3-[(6-氨磺酰基-2-吡啶基)氨基]丙基]吡咯烷-1-甲酸叔丁酯(7g,16.97mmol)进行手性分离,得到作为第一个洗脱峰的(4s)-2,2-二甲基-4-[3-[(6-氨磺酰基-2-吡啶基)氨基]丙基]吡咯烷-1-甲酸叔丁酯(3.4481g,99%)。esi-ms m/z计算值412.21442,实验值413.2(m+1)
+
;保留时间:0.63分钟(lc方法a)。
[0299]
部分c:(14s)-8-[3-(2-{二螺[2.0.2.1]庚-7-基}乙氧基)-1h-吡唑-1-基]-12,12-二甲基-2λ6-硫杂-3,9,11,18,23-五氮杂四环[17.3.1.111,14.05,10]二十四碳-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮(化合物ii)的合成
[0300][0301]
步骤1:(4s)-4-[3-[[6-[[2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-羰基]氨磺酰基]-2-吡啶基]氨基]丙基]-2,2-二甲基-吡咯烷-1-甲酸叔丁酯
[0302][0303]
向2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酸(5.2g,14.45mmol)于四氢呋喃(100ml)中的溶液中添加羰基二咪唑(2.8g,16.51mmol),并且将混合物在环境温度下搅拌1小时。向该混合物中添加含(4s)-2,2-二甲基-4-[3-[(6-氨磺酰基-2-吡啶基)氨基]丙基]吡咯烷-1-甲酸叔丁酯(6.0g,14.54mmol)于四氢呋喃(15ml),随后添加1,8-二氮杂双环[5.4.0]十一碳-7-烯(6.5ml,43.47mmol),并且将混合物在环境温度下搅拌16小时。将反应物用水(150ml)稀释并用盐酸水溶液(15ml,6m,90.00mmol)使混合物酸化。将混合物用乙酸乙酯(300ml)萃取,并分离有机相。将有机相用盐水洗涤,经硫酸镁干燥,经硅藻土过滤并真空浓缩,得到白色沉淀。将沉淀与乙腈一起制浆,并且使用中型玻璃料通过过滤收集固体并用乙腈洗涤。将滤液真空浓缩,得到黄色油状物。将粗制油状物用乙腈和一些n-甲基-2-吡咯烷酮稀释,并且在415g反相c
18
柱上用50%-100%乙腈/水进行洗脱来色谱分析,得到(4s)-4-[3-[[6-[[2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-羰基]氨磺酰基]-2-吡啶基]氨基]丙基]-2,2-二甲基-吡咯烷-1-甲酸叔丁酯(4.5g,41%)。esi-ms m/z计算值753.30756,实验值754.4(m+1)+;保留时间:3.79分钟(lc方法d)。
[0304]
步骤2:2-氯-n-[[6-[3-[(3s)-5,5-二甲基吡咯烷-3-基]丙基氨基]-2-吡啶基]磺酰基]-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酰胺(三氟乙酸盐)
[0305][0306]
向(4s)-4-[3-[[6-[[2-氯-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-羰基]氨磺酰基]-2-吡啶基]氨基]丙基]-2,2-二甲基-吡咯烷-1-甲酸叔丁酯(5.9g,7.821mmol)于二氯甲烷(30ml)和甲苯(15ml)中的溶液中添加三氟乙酸(6.0ml,77.88mmol),并且将混合物在环境温度下搅拌18小时。将溶剂真空去除,浴温设为45℃,得到浓稠黄色油状物。将油状物用甲苯(125ml)稀释并将溶剂真空去除,浴温设为45℃。将油状物用甲苯稀释并将溶剂真空去除,得到浓稠粘性黄色油状物2-氯-n-[[6-[3-[(3s)-5,5-二甲基吡咯烷-3-基]丙基氨基]-2-吡啶基]磺酰基]-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酰胺(三氟乙酸盐)(6.0g,100%),其不经进一步纯化即用于下一步骤中。esi-ms m/z计算值653.2551,实验值654.3(m+1)+;保留时间:2.6分钟(lc方法b)。
[0307]
步骤3:(14s)-8-[3-(2-{二螺[2.0.2.1]庚-7-基}乙氧基)-1h-吡唑-1-基]-12,12-二甲基-2λ6-硫杂-3,9,11,18,23-五氮杂四环[17.3.1.111,14.05,10]二十四碳-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮(化合物ii)
[0308][0309]
向2-氯-n-[[6-[3-[(3s)-5,5-二甲基吡咯啶-3-基]丙基氨基]-2-吡啶基]磺酰基]-6-[3-(2-二螺[2.0.2.1]庚-7-基乙氧基)吡唑-1-基]吡啶-3-甲酰胺(三氟乙酸盐)(6.0g,7.810mmol)于nmp(140ml)中的溶液中添加碳酸钾(5.3g,38.35mmol)。将混合物用氮气吹扫5分钟。然后将混合物在150℃下加热22小时。将反应混合物冷却至室温并添加到水(300ml)中,得到灰白色固体沉淀。用盐酸水溶液(12ml,6m,72.00mmol)小心地使混合物酸化,得到泡沫状浆液。使用中型玻璃料通过过滤收集固体。将湿滤饼溶解于乙酸乙酯(500ml)中,并用200ml盐水洗涤。水相略微浑浊,因此用少量6n盐酸使其酸化并返回至有机相。分离水相,并将有机相经硫酸镁干燥,过滤并真空浓缩,得到浅黄色油状物。将该粗产物用乙腈稀释,并在415g c
18
反相柱上用50%-100%乙腈/水进行洗脱来进行色谱分析。分离出呈乳油色泡沫状物的产物。将泡沫在真空中在45℃下干燥48小时,得到(14s)-8-[3-(2-{二螺[2.0.2.1]庚-7-基}乙氧基)-1h-吡唑-1-基]-12,12-二甲基-2λ6-硫杂-3,9,11,18,23-五氮杂四环[17.3.1.111,14.05,10]二十四碳-1(22),5,7,9,19(23),20-己烯-2,2,4-三酮(化合物ii)(3.32g,68%)。1h nmr(400mhz,二甲基亚砜-d6)δ12.48(s,1h),8.20(d,j=2.8hz,1h),7.81(d,j=8.2hz,1h),7.57(dd,j=8.5,7.2hz,1h),7.05(d,j=7.1hz,1h),6.97(d,j=8.5hz,1h),6.91(d,j=8.2hz,1h),6.71(d,j=8.5hz,1h),6.08(d,j=2.7hz,1h),4.21(td,j=6.7,1.3hz,2h),3.92(d,j=12.0hz,1h),3.16(s,1h),2.95(d,j=13.3hz,1h),2.78-2.66(m,1h),2.07(s,1h),1.92-1.72(m,4h),1.60(s,6h),1.51(s,3h),
1.47(t,j=6.5hz,1h),1.31(q,j=12.2hz,1h),0.89-0.77(m,4h),0.69-0.61(m,2h),0.53-0.45(m,2h).esi-ms m/z计算值617.27844,实验值618.4(m+1)
+
;保留时间:10.29分钟(lc方法f)。
[0310]
实例5:化合物ii(游离形式)形式a
[0311]
向反应器配备顶置式搅拌器、回流冷凝器、n2鼓泡管线和出口以及温度探针。将(14s)-8-溴-12,12-二甲基-2λ
6-硫杂-3,9,11,18,23-五氮杂四环[17.3.1.1
11,14
.05,
10
]二十四碳-1(23),5,7,9,19,21-六烯-2,2,4-三酮(120g,86%w/w,含有ipac,[103.2g(14s)-8-溴-12,12-二甲基-2λ
6-硫杂-3,9,11,18,23-五氮杂四环[17.3.1.1
11
,
14
.05,
10
]二十四碳-1(23),5,7,9,19,21-六烯-2,2,4-三酮],0.21mol,1当量)、3-(2-(二螺[2.0.24.13]庚-7-基)乙氧基)-1h-吡唑(42.6g,0.21mol,1当量)、325目k2co3(63.4g,0.46mol,2.2当量)、cui(3.3g,17.2mmol,0.083当量)和buoac(740ml)的混合物装入到反应器中。将混合物在环境温度下搅拌。然后将dmf(300ml,2.9vol)和
[0312]
n,n
′‑
二甲基环己烷-1,2-二胺(14.6g或16.2ml,0.1mol,0.49当量)装入到反应器中,并且将混合物用三次n2/真空/n2循环吹扫。然后将混合物加热至120℃持续4小时,然后使其冷却至环境温度。逐滴添加10%w/v草酸水溶液(860ml,0.96mol,4.6当量),并且将混合物搅拌至少1小时。然后将混合物过滤以去除悬浮固体。用(2x 120ml)洗涤去除的固体。分离来自滤液的各层。将有机层用8%w/v柠檬酸三钠水溶液(600ml)洗涤。根据需要添加盐水以帮助相分离。将有机层用1:1v/v水/盐水(400ml)洗涤。经由硅藻土垫过滤有机层。用ipac(150ml)洗涤过滤垫。将滤液浓缩,然后添加800ml 1-proh(7.8vol)并将混合物浓缩。将此步骤再重复一次。添加甲苯(800ml)并浓缩混合物。将此步骤再重复一次,得到浓稠浆液。将粗制混合物浓缩至300ml(2.9vol)的甲苯体积。在搅拌浆液过夜之后,通过过滤收集固体,并用甲苯(2x 100ml,0.97vol)洗涤固体。将固体在50℃下在真空用氮气排放干燥,直到干燥损失不超过1.0%,得到呈白色/类白色固体的化合物ii(107.0g,83%,94.5%(auc)hplc纯度。
[0313]
再结晶:将化合物ii形式a[22.2g,94.6%(auc)化合物ii形式a悬浮于甲苯(440ml,基于化合物ii形式a的20vol)中并将混合物加热至回流。在回流下保持nlt 2小时后,历经8小时使混合物冷却至环境温度。在环境温度下搅拌过夜后,通过过滤收集固体,用甲苯(40ml,1.8vol)洗涤该固体。将固体在50℃下在真空用氮气排放干燥,直到干燥损失不超过1.0%,得到呈白色/类白色固体的化合物ii形式a(18.8g,84%,96.8%(auc)hplc纯度)。
[0314]
第二次再结晶:将化合物ii形式a[17.5g,97.0%(auc)化合物ii形式a]悬浮于甲苯(350ml,基于化合物ii形式a的20vol)中并将混合物加热至回流。在回流下保持不少于2小时后,历经8小时使混合物冷却至环境温度。在环境温度下搅拌过夜后,通过过滤收集固体,用甲苯(40ml,1.8vol)洗涤该固体。将固体在50℃下在真空用氮气排放干燥,直到干燥损失不超过1.0%,得到呈白色/类白色固体的化合物ii形式a(游离形式)(15.7g,89%,98.4%(auc)hplc纯度)。
[0315]
化合物ii游离形式a在室温下水活性≤0.95时是最稳定的多晶形式。
[0316]
a.x射线粉末衍射
[0317]
使用配备有vantec-1检测器的bruker advance,在室温下以反射模式获取xrpd图
案。在硅样品架上从3
°
2θ至40
°
2θ以连续模式对样品进行分析,其中步长为0.0144531
°
并且每步时间为0.25秒。以15rpm旋转样品。化合物ii(游离形式)形式a的xrpd衍射图提供于图1中并且xrpd数据总结于下表5中。
[0318]
表5:化合物ii(游离形式)的结晶形式a的xrpd信号
[0319][0320][0321]
b.单晶阐明
[0322]
从丙酮/庚烷中生长具有化合物ii(游离形式)形式a结构的单晶。在配备有mo k
α
辐射和ccd检测器的bruker衍射仪上在298k下获取x射线衍射数据。使用shelx程序对结构进行解析和精修(sheldrick,g.m.,acta cryst.,(2008)a64,112-122),并且结果汇总于下表6中。
[0323]
表6:化合物ii(游离形式)形式a的单晶阐明
[0324][0325][0326]
c.固态nmr
[0327]
1.固态nmr实验(适用于化合物ii的所有结晶形式):
[0328]
使用配备有bruker-biospin 4mm hfx探针的bruker-biospin 400mhz宽口径光谱仪。将样品装填至4mm zro2转子中并在魔角旋转(mas)条件下以通常设定为12.5khz的旋转速度旋转。使用1h mas t1饱和恢复弛豫实验测量质子弛豫时间以设定
13
c交叉极化(cp)mas实验的适当循环延迟。将碳cpmas实验的cp接触时间设定为2毫秒。采用具有线性斜坡(50%至100%)的cp质子脉冲。在外部参照样品(甘氨酸)上使碳hartmann-hahn匹配优化。使用tppm15去耦序列在大约100khz的场强下利用质子去耦记录碳谱。
[0329]
2.化合物ii(游离形式)形式a的固态nmr
[0330]
化合物ii(游离形式)形式a的固态
13
c nmr数据提供于图2中并总结于下表7中。
[0331]
表7:化合物ii(游离形式)形式a的固态nmr
[0332]
[0333][0334]
d.差示扫描量热法分析
[0335]
使用ta discovery差示扫描量热仪(ta instruments,new castle,de)进行dsc。仪器用铟校准。将大约1-10mg的样品称重至密封盘中,使用带有一个孔的盖子将其压接。以10℃/分钟的加热速率从25℃至300℃扫描dsc样品。收集数据并通过trios分析软件(ta instruments,new castle,de)进行分析。热谱图显示在约227℃处的单一熔融吸热峰。
[0336]
实例6:化合物ii钙盐水合物形式a
[0337]
化合物ii钙盐水合物形式a是动力学上最有利的钙盐水合物形式,提供了比其它钙盐水合物形式更高的溶解、溶解度和暴露。
[0338]
化合物ii钙盐水合物形式a是通过向0.2mmol的化合物ii(游离形式)形式a和0.1mmol的ca(ome)2干粉中装入约45mg/ml的ipa并掺入约10%的水并加热至70℃来制备的。最初,所有固体都溶解。5分钟后,白色固体沉淀出。将所得浆液在室温下搅拌4天。通过真空过滤将固体分离为化合物ii钙盐水合物形式a并在真空下在40℃下干燥过夜(约78%的分离产率)。
[0339]
制备化合物ii钙盐水合物形式a的替代方法利用装有63ml ipa和7ml水的10g的化合物i(游离形式a)。将浆液加热至55-65℃。向混合物中装入1.1当量的naoh。搅拌混合物直至溶液变均质为止。然后将溶液冷却至25℃并用0.1g的化合物ii氯化钠水合物形式a接种。将浆液搅拌18小时。然后将溶液加热至45℃。将浆液用0.1g的化合物ii钙盐水合物形式a接种。在5小时的时间段内添加0.55当量cacl2、9ml ipa和1ml水的溶液。将所得浆液搅拌2小时。在5小时内将浆液冷却至20℃。通过真空过滤收集所得固体,并将所得湿滤饼用50ml水洗涤。使洗涤的湿滤饼风干1小时。将风干的湿滤饼转移到45℃下的真空烘箱中,轻微氮气排放20小时,得到结晶化合物ii钙盐水合物形式a(8.5g,82%分离产率)。
[0340]
a.x射线粉末衍射:
[0341]
使用配备有vantec-1检测器的bruker advance,在室温下以反射模式获取xrpd图案。在硅样品架上从3
°
2θ至40
°
2θ以连续模式对样品进行分析,其中步长为0.0144531
°
并且每步时间为0.25秒。以15rpm旋转样品。化合物ii钙盐水合物形式a的xrpd衍射图示出于图3中并总结于表8中。
[0342]
表8:结晶化合物ii钙盐水合物形式a的xrpd信号
[0343][0344][0345]
b.单晶阐明
[0346]
通过将1mg的化合物ii钙盐水合物形式a溶解于350μl的二氯乙烷/乙醇的90/10混合物中使具有化合物ii钙盐水合物形式a结构的晶体生长并且然后用戊烷蒸气扩散几天。在配备有cu k
α
辐射和ccd检测器的bruker衍射仪上在100k和298k两者下获取x射线衍射数据。使用shelx程序对结构进行解析和精修(sheldrick,g.m.,acta cryst.,(2008)a64,112-122),并且结果汇总于下表9中。
[0347]
表9:化合物ii钙盐水合物形式a的单晶阐明
[0348][0349]
c.固态nmr
[0350]
化合物ii钙盐水合物形式a的固态
13
c nmr谱提供于图4中并总结于表10中。
[0351]
表10:化合物ii钙盐水合物形式a的固态nmr
[0352]
峰编号化学位移[ppm]
±
0.2强度[相对]1178.327.02165.238.83158.227.64155.832.75153.120.56150.929.97143.442.18136.841.39127.931.510116.327.411114.648.212112.147.41398.627.41493.641.21569.533.01668.617.01763.855.61857.743.11951.850.22042.934.22137.249.92231.216.92329.648.12426.4100.02520.877.4
2617.065.8277.844.7285.348.8292.618.7
[0353]
d.差示扫描量热法分析:
[0354]
使用ta instruments dsc q2000获得dsc热谱图。以10℃/分钟将样品从30℃加热至350℃。热谱图显示在约223℃处的吸热峰。
[0355]
实例7:化合物ii钙盐水合物形式d
[0356]
化合物ii钙盐水合物形式d在某些条件下,如在乙醇和水的混合物中,是钙盐水合物的最稳定形式。
[0357]
向大约25mg的化合物ii钙盐水合物形式a中装入0.5ml的etoh:水67:33w/w。将浆液加热至65℃,持续8天。通过真空过滤收集的所得固体是化合物ii钙盐水合物形式d。
[0358]
可替代地,化合物ii钙盐水合物形式d由89g的装有1080ml ipa和120ml水的化合物ii钠水合物形式a制备。将浆液加热至55-65℃。向浆液中装入18g的化合物ii钙盐水合物形式d晶种。湿磨浆液,同时在5小时的时间段内添加0.55当量cacl2、81ml ipa和9ml水的溶液。使湿磨运行直至x射线粉末衍射证实浆液全部为化合物i钙盐水合物形式d为止。通过真空过滤收集所得固体并用350ml水洗涤湿滤饼。使洗涤的湿滤饼风干1小时。将风干的湿滤饼转移到45℃下的真空烘箱中,轻微氮气排放20小时,得到结晶化合物ii钙盐水合物形式d(83.15g,90.6%分离产率)。
[0359]
化合物ii钙盐水合物形式d在室温至60℃的水活性为0.1-0.95的ipa/水中是最稳定的多晶形式。
[0360]
a.x射线粉末衍射:
[0361]
使用配备有密封管源和pixcel 1d medipix-2检测器的panalytical empyrean系统(malvem panalytical inc,westborough,massachusetts),在室温下以反射模式记录x射线粉末衍射(xrpd)谱。利用铜辐射在45kv的电压和40ma的电流下运行x射线发生器。将粉末样品置于背面填充的样品架中并装载至仪器中。在约3
°
2θ至约40
°
2θ的范围内扫描样品,其中步长为0.0131303
°
并且每步为49.725秒。化合物i钙盐水合物形式d的xrpd衍射图示出于图5中并总结于表11中。
[0362]
表11:结晶化合物ii钙盐水合物形式d的xrpd信号
[0363]
[0364][0365]
b.单晶阐明
[0366]
晶体选自化合物ii钙盐水合物形式d在乙醇/水中的接种过程。在配备有cu ka辐射(1=1.5478,由rigaku mm007hf旋转阳极提供)和cmos检测器的bruker衍射仪上在100k下获取x射线衍射数据。使用shelx程序对结构进行解析和精修(sheldrick,g.m.,acta cryst.,(2008)a64,112-122),并且结果汇总于表12中。
[0367]
表12:化合物ii钙盐水合物形式d的单晶阐明
[0368][0369]
c.固态nmr:
[0370]
化合物i钙盐水合物形式c的固态
13
c nmr谱提供于图6中并总结于表13中。
[0371]
表13:化合物ii钙盐水合物形式d的固态nmr
[0372]
[0373][0374]
[0375]
d.差示扫描量热法分析
[0376]
使用ta discovery差示扫描量热仪(ta instruments,new castle,de)进行dsc。仪器用铟校准。将大约1-10mg的样品称重至密封盘中,使用带有一个孔的盖子将其压接。以10℃/分钟的加热速率从25℃至300℃扫描dsc样品。收集数据并通过trios分析软件(ta instruments,new castle,de)进行分析。热谱图显示在约182℃和约208℃处的多个吸热峰。
[0377]
实例8:250mg的化合物i的功效数据
[0378]
在临床试验中,在采用依伐卡托稳定治疗的门控突变受试者中进行了关于在12周时swcl的绝对变化的比较研究。通常认为化合物i是安全的并且以150mg和250mg良好耐受,持续12周。250mg剂量证明与依伐卡托基线相比在第12周时swc1改善,而150mg剂量的化合物1证明与依伐卡托基线相比swcl降低。
[0379][0380]
实例9:示例性片剂调配物的制备
[0381]
对表15中的颗粒内组分(化合物i喷雾干燥分散体(sdd)、化合物ii钙盐水合物形式d、化合物iii sdd、微晶纤维素和交联羧甲基纤维素钠)进行称重并通过筛网筛分并置于箱式搅拌机中。将这些组分共混并合并以制备颗粒内粉末共混物。使用辊压实机将颗粒内粉末共混物干法制粒并且然后研磨成颗粒。对颗粒外微晶纤维素进行称重,通过筛网并在箱式搅拌机中与磨碎的颗粒共混。对硬脂酸镁进行称重并筛分,然后添加到箱式搅拌机中并共混。使用动力辅助旋转压片机对共混的组分进行压缩以制备具有所需核心重量和硬度的片剂。然后将片剂放入包衣机中以添加非功能性包衣。
[0382]
表15.包括125mg化合物i、10.6mg化合物ii钙盐水合物形式d和50mg化合物iii的示例性片剂调配物。
[0383][0384]
其它实施例
[0385]
前述论述仅公开并描述了本公开的示例性实施例。所属领域的技术人员将从此类论述以及从附图和权利要求书中容易地认识到,在不脱离如以下权利要求书中限定的本公开的精神和范围的情况下,可在其中进行各种改变、修改和变化。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1