一种基于势能面匹配的材料力场拟合方法
1.本发明涉及材料信息学技术领域,尤其涉及一种基于势能面匹配的材料力场拟合方法。
背景技术:
2.基于量子化学与分子力学的微纳米跨尺度集成计算方法,以及逐步替代传统实验室中高成本的“试错法”,将其应用于lammps等经典牛顿力学框架下分子动力学全原子水平模型,可以精确描述微结构变化、可视化揭示互相作用机理、预测材料的热力学特性、输运性质及本构特性,有助于从原子层面指导材料的宏观性能调控及高通量筛选。
3.纤维是横向尺度微观、轴向尺度宏观的固体结构材料,具有柔韧性、纤细和长径比高的特点,广泛应用于纺织结构及纤维增强复合材料中,在工程中具有可设计性和巨大的应用潜力,对于碳纤维、玻璃纤维、芳纶纤维等常见增强纤维,其屈服强度,断裂强度以及断裂延伸率为评价增强效果的重要指标。
4.分子动力学将共价键处理为量子效应的简谐振子,并通过力场实现体系立体构型势能面的经验拟合,因此力场的质量决定了仿真结果的精度与指导效果。常见的经典力场包括cff91、amber、charmm、cvff;热规划力场包括uff、valbond;二代力场包括pcff、mmff、compass;上述力场在研究中都有不错的成效,但是很多参数未公开或缺乏有效标定,难以准确评估参数的可靠性。
5.材料拉伸变化可分解为键相互作用(键势)和非键相互作用(对势),其中,键相互作用包括键伸缩势、键角弯曲能、二面角扭转能、离面角扭转能以及更为复杂的耦合效应的交叉项,非键相互作用包括范德华作用、库伦相互作用、氢键等,键伸缩势在体系中往往占主导作用。传统的简谐键势无法描述断键过程,而简单地将原子共价键半径和作为断键判据精度过低。
6.专利文献cn109994158a公开了一种基于强化学习构建分子反应力场的系统及方法,包括输出输入模块,参数与配置模块、分子动力学接口模块、环境设置模块、优化模块;通过封装预先训练获得的深度学习模型,完成分子力场的构建。
7.该系统通用性很强,但是整个系统非常依赖于预先训练的模型,但是该模型是基于采样的方式进行判定,通过迭代筛选出最符合要求的力场,运算压力过大且没有针对性的参数使得输出力场文件模拟结果效果并不佳。
8.专利文献cn110473596a公开了一种基于势能面扫描的铝电解熔盐体系力场拟合方法:
9.1)通过计算熔盐体系中任意两离子键的势能面;
10.2)计算熔盐中离子的电荷和静电互相作用势能;对离子间的静电互相作用势能进行分解,得到buckingham势能;
11.3)采用非线性拟合方法拟合buckingham势函数,得到适用于熔盐体系分子动力学的势参数;
12.4)根据获得的势参数进行力学测试,计算熔盐体系的离子结构和输运性质。
13.该方法只适用于离子间非键相互作用的力场拟合,并不适合关于强度方面的高精度力学性能分析。
技术实现要素:
14.为了解决上述问题,本发明提供了一种基于势能面匹配的材料力场拟合方法,该方法通过引入无穷远处具有有限势阱深度的morse键伸缩势参数,使得最终仿真模拟关于屈服强度,断裂强度以及断裂延伸率的测试结果更加符合实际,进而能更好的了解纤维材料的物理力学性能与构效关系。
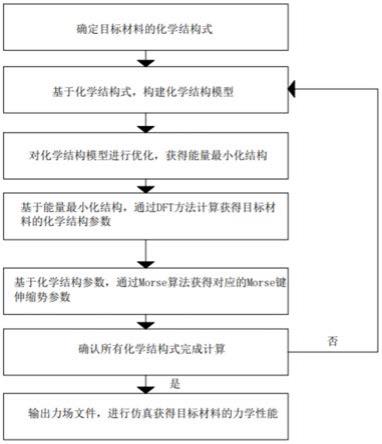
15.一种基于势能面匹配的材料力场拟合方法,包括:
16.s1根据目标材料,获取对应的化学结构式;
17.s2基于s1中化学结构式的原子杂化状态,构建关于所述化学结构式中连接键的化学结构模型;
18.s3对s2中构建获得的化学结构模型,进行结构初步优化,获得能量最小化结构;
19.s4基于s3中获得的能量最小化结构,通过dft方法计算获得目标材料的化学结构参数;
20.s5基于s4中获得的化学结构参数,通过morse算法获得morse键伸缩势参数,并保存在分子模拟文件中;
21.s6重复上述s2-s5,直至完成所有化学结构模型的计算,并将所有分子模拟文件集成进力场文件;
22.s7基于s6中获得的力场文件,进行分子模拟仿真,计算目标材料的力学性能。
23.本发明提供的方法针对以拉伸屈服强度、断裂强度以及断裂延伸率为主要评估指标的材料。
24.优选的,所述目标材料为纤维材料。
25.具体的,所述s2中化学结构模型包括团簇结构模型与断键后自由基片段模型。
26.具体的,所述s3中结构初步优化是基于mmffs分子力场,结合最快速下降法与共轭梯度法对化学结构模型进行优化,便于后续的量化计算。
27.具体的,所述s4中的化学结构参数包括平衡态键长、电子能、平衡态振动频率以及对应的振动波数。
28.具体的,所述s4的具体过程为:
29.s4.1通过久期方程对能量最小化结构进行振动频率分析,获得平衡态振动频率;
30.s4.2结合s4.1获得平衡态振动频率的振型图,通过红外线光谱分析获得对应的振动波数;
31.s4.3通过设置泛函,基组,静电荷数与自旋转多重态参数,通过密度泛函理论对能量最小化结构进行波函数分析,获得对应的电子能以及平衡态键长。
32.具体的,所述s5中的morse键伸缩势参数包括键解离势阱深度、平衡态键长以及控制平衡位置势能曲线曲率。
33.具体的,所述s5的具体过程为:
34.s5.1通过团簇结构模型与断键后自由基片段模型之间的电子能差值,计算键解离
势阱深度:
[0035][0036]
其中,h为普朗克常数,c为光速,d
ν
为波长单位表达的键解离势阱,为第i个自由基片段摩尔焓,为团簇模型摩尔焓,δec为温度修正项,r为理想气体常数,t为温度,e
el,fr,i
为断键后自由片段的电子能,e
el,cl
为团簇结构的电子能;
[0037]
s5.2控制平衡位置势能曲线曲率计算:
[0038]
α=(8π2cμνeχ/h)
1/2
=0.2454(mνeχ)
1/2
=0.1227νe(m/d
ν
)
1/2
[0039]
其中m=m1m2/(m1+m2),m为双原子系统的约化质量,m1与m2分别为双原子中每个原子的摩尔质量,χ为非谐性常数,ve为平衡态振动频率对应波数。
[0040]
所述目标材料的力学性能包括屈服强度、断裂强度以及断裂延伸率。
[0041]
与现有技术相比,本发明的有益效果:
[0042]
(1)在力场拟合的过程中,通过非线性morse型指数函数拟合,获得更具有物理意义的morse键伸缩势参数,通过包含morse键伸缩势参数的力场文件构建模型,其模型能更好的体现材料的物理力学性能与构效关系。
[0043]
(2)相较于传统方法直接将共价键半径之和作为断键判断,本发明采用非线性morse型指数函数拟合方法,来规避包含电子密度积分运算的键级反应力场,从而降低了运算时间。
[0044]
(3)本发明方法除了基础目标材料的化学结构以外,需要的实验数据少,从而减少了时间成本,同时也节约了实验测试成本。
附图说明
[0045]
图1为本发明提供的材料力场拟合方法的流程示意图;
[0046]
图2为实施例中ch3cn团簇模型的传统简谐键伸缩势曲线与morse键伸缩势曲线对比图;
[0047]
图3为实施例中传统方法与本发明提供的方法之间仿真结果的对比图;
[0048]
图4为实施例中通过本发明提供的方法生成的纤维预氧丝模型非平衡态拉伸俯视图;
[0049]
图5为实施例中传统方法生成的纤维预氧丝模型非平衡态拉伸俯视图。
具体实施方式
[0050]
如图1所示,一种基于势能面匹配的材料力场拟合方法,具体步骤为:
[0051]
s1根据目标材料,获取对应的化学结构式:以包含c/h/o/n元素的纤维预氧丝模型体系为例该体系涉及的化学结构式以及对应的键类型,如表1所示:
[0052]
表1实施例中包含的化学结构式以及对应的键类型
[0053]
团簇模型键类型ch3cnc
sp-c
sp3
hcnc
sp-n
sp
hc(o)hc
sp2-o
sp2
ch4c
sp3-hc6h5nh2c
ring-n
sp3
c6h5ohc
ring-o
sp3
c6h6c
ring-c
ring
c6h6c
ring-hc5h5nc
ring-n
ring
nh3n
sp3-hch3oho
sp3-hc2h6c
sp3-c
sp3
[0054]
s2根据s1中获取的化学结构式,基于所述化学结构式的原子杂化状态,构建关于所述化学结构式中连接键的化学结构模型:
[0055]
以c
sp-c
sp3
键为例,基于杂化形式补齐原子,根据团簇模型ch3cn通过avogadro分子建模软件中建立化学结构模型,其中包含团簇模型ch3cn与断键后自由基片段模型
·-ch3及
·-cn。
[0056]
s3对s2中构建获得的化学结构模型,进行结构初步优化,获得能量最小化结构:基于mmffs分子力场,结合最速下降法和共轭梯度法组合优化,将s2中的化学结构模型优化为能量最小化结构,在extensions内生成orca量化计算软件的inp输入文件。
[0057]
s4基于s3中获得的能量最小化结构,通过dft方法计算获得目标材料的化学结构参数:编辑量化计算输入文件的关键词,选择如交换关联泛函数中含有20%hf成分的b3lyp泛函;根据最终测试精度需要,选择基组类型如def2-svp劈裂价键基组,选用关键词“opt”进行结构优化,选用关键词“freq”采用久期方程实现振动频率分析;同时设置净电荷数及自旋多重态参数,在软件orca中对团簇模型ch3cn展开密度理论分析。
[0058]
最终通过软件chemcraft可视化输出文件,最优团簇模型ch3cn的结构电子能e
el,c
l=-8.3194*104kcal/mol,平衡位置键长结合振型图以及红外光谱分析经验值确定团簇模型ch3cn的振动波数νe=1388.59cm-1
。
[0059]
采用相同泛函、基组以及关键词“sp”,通过软件orca对断键后自由基片段模型
·-ch3及
·-cn展开密度泛函理论分析,最终通过软件chemcraft可视化输出文件,最优断键后自由基片段模型
·-ch3及
·-cn的结构电子能分别为-2.4961*104kcal/mol与-5.8102*104kcal/mol。
[0060]
s5基于s4中获得的化学结构参数,通过morse算法获得morse键伸缩势参数,并保存在分子模拟文件中:
[0061]
原始morse公式:
[0062][0063]
其中,r为双原子间距离,re为平衡位置键长,d为键解离势阱深度,α为控制平衡位置势能曲线曲率。
[0064]
基于多次参数变换、广义laguerre多项式及分数阶微分归一化积分算法得到振动能的离散表达式:
[0065][0066]
其中,χ为非谐性常数,νe为平衡态振动频率对应波数,非谐性常数为键解离势阱深度与振动波数或频率的函数。
[0067]
振动波数可通过dft振动分析以及光谱分析获得,而键解离势阱深度可以通过dft方法求解团簇结构模型与断键后自由基片段模型之间的电子能差值进行计算,:
[0068][0069]
其中,h为普朗克常数,c为光速,d
ν
为波长单位表达的键解离势阱,为第i个自由基片段摩尔焓,为团簇模型摩尔焓,δec为温度修正项,r为理想气体常数,t为温度,e
el,fr,i
为断键后自由基片段的电子能,e
el,cl
为团簇结构的电子能;由于双原子体系键被重复计算,最终的势能参数d取de值的一半,最终得到键解离势阱深度为65.80kcal/mol。
[0070]
控制平衡位置势能曲线曲率计算:
[0071]
α=(8π2cμνeχ/h)
1/2
=0.2454(mνeχ)
1/2
=0.1227νe(m/d
ν
)
1/2
[0072]
其中m=m1m2/(m1+m2)双原子系统的约化质量,χ为非谐性常数,ve为平衡态振动频率对应波数;得到平衡位置势能曲线曲率为
[0073]
将上述获得的参数,带入拟合后的morse型键伸缩势表达式:
[0074]emorse
=65.80*[1-e-1.945*(r-1.459)
]2[0075]
基于拟合后的morse型键伸缩势绘制曲线图,并与传统方法获得的曲线图进行比对:
[0076]
结果如图2所示,说明本方法的拟合结果在平衡位置(即图中原子间距为1.5附近)与传统简谐键伸缩势曲线具有良好的一致性,同时在原子距离较远时出现了断键情况(即图中原子距离为2.5附近),相较于传统简谐键伸缩势曲线比,更加符合实际情况,图中虚线为传统简谐键伸缩势曲线,实线为morse键伸缩势曲线。
[0077]
s6重复上述s2-s5,直至完成所有化学结构式的计算,并将所有分子模拟文件集成
进力场文件:
[0078]
将参数写入lammps的in及data文件中,
[0079]
其中in文件格式为:bond_style morse
[0080]
data文件格式为:bond coeffs${bond#}65.80 1.945 1.459。
[0081]
其中本实施例的纤维预氧丝模型所有键类型及拟合参数,如表2所示:
[0082][0083]
s7基于s6中获得的力场文件,进行分子模拟仿真,计算目标材料的力学性能,其中力学性能包括屈服强度、断裂强度以及断裂延伸率。
[0084]
根据仿真模拟结果与传统的方法进行比较,如图3所示,图中虚线为传统简谐键伸缩势力场曲线,实线为morse键伸缩势力场曲线:根据对比图可知传统简谐键伸缩势应力随应变增大线性增大,且在9%名义应变下未能观察到骤降情况(即未出现断键点);而morse键伸缩势应力应变曲线图则在9%名义应变时出现了骤降情况(即出现断键点),其过程更加符合实际情况。
[0085]
如图4、5所示,图中黑色加粗长条为原子间出现的纳米孔洞或微裂纹,本发明方法生成的模型在仿真过程中,当名义应变达到9%时能明显观察到断键位置,即符合实际情况;而传统方法并没有出现明显的纳米孔洞或微裂纹,不符合实际情况。
[0086]
最后根据仿真结果可知,本实施例中的纤维预氧丝断裂强度为7.95gpa,断裂延伸率为8.83%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1