一种基于生物子结构预测药物-靶标相互作用的深度学习方法
1.本发明涉及生物信息与深度学习领域,具体涉及一种基于生物子结构预测药物-靶标相互作用的深度学习方法。
背景技术:
2.药物-靶标相互作用(drug target interaction,dti)具体是指细胞内的特定生物大分子(称为“靶标”,大多数是蛋白质,例如酶、离子通道等)以适当的化学特性和亲和力与药物分子结合的过程,是药物治疗疾病的内在机制,因此药物-靶标相互作用是药物发现和开发的基础。目前,基于体外实验的相互作用分析方法面临着时间周期长和金钱消耗大的问题,近些年来,随着深度学习在生物信息学的各个领域的发展和成就,其在药物-靶标相互作用预测任务上也表现出了巨大的研究潜力,利用深度学习的方法预测药物-靶标相互作用的解决方案,能够加快药物发现的速度、降低研究成本。
3.基于深度学习的药物-靶标相互作用预测方法通常分为特征提取和分类预测两步,由于药物分子通常由smiles(simplified molecular input line entry system)序列、分子指纹和分子图进行表示,而靶标由氨基酸序列进行表示,所以特征提取常常借助语言模型和图神经网络围绕着生物信息、拓扑结构以及物化属性方向进行。在特征提取阶段,借鉴语言模型的方法,往往以一个药物和靶标的序列表示作为输入,借助一些语言模型分别对药物和靶标序列进行编码,例如word2vec,doc2vec和transformer等,从而获得药物和靶标的语义特征表示;一些研究以药物的分子图作为输入,采用gnn,gcn等图神经网络提取药物的拓扑结构特征作为其特征表示。在分类预测中,将以上药物和靶标的特征进行拼接,作为分类器的输入,经过多层全连接网络学习相互作用特征,从而预测两者之间是否会发生相互作用。这些方法仍存在着以下几点局限性:
4.1)提取药物分子特征的粒度大都是原子级别或者以整个分子信息和靶标信息作为其输入,这种处理方式不具备药理反应理论依据,还可能会引入噪音,在不同分布数据集上的效果差异较大,并且使得预测缺乏可解释性。
5.2)部分研究借助自然语言处理等技术,提取药物的子结构进行预测,这些方法大多数依靠数据统计,缺乏生物学领域理论支持。
技术实现要素:
6.本发明的目的是:为缓解现有药物-靶标相互作用预测技术中存在的局限性,受药效团和药理学知识启发,即药物分子中对药物活性起重要贡献的往往是一些药物基团和子结构,并且药物分子支链的长度和种类等也会影响分子的亲和力,进而影响药物和靶标间的相互作用而提出的一种基于生物子结构的药物-靶标相互作用预测的深度学习方法,该方法以药物分子、靶标及其相互作用数据为研究对象,提取出包括分子支链、常见子结构和逆合成片段在内的多种药物子结构,同时借助自然语言处理技术提取出靶标子结构,在此
基础上构建深度学习模型对药物-靶标相互作用进行建模,学习更全面的药效性质,获得高层次、多维度的药理特征,提高预测的准确率,加速药物开发。
7.实现本发明目的的具体技术方案是:
8.一种基于生物子结构预测药物-靶标相互作用的深度学习方法,包括以下步骤:
9.步骤a:输入一个药物的规范化smiles序列d和靶标氨基酸序列t;
10.步骤b:分别对药物的规范化smiles序列d和靶标氨基酸序列t进行子结构提取,具体包括:
11.1)对于药物的规范化smiles序列d,使用bcm方法提取药物的子结构,包括:
12.1.1)首先从药物的规范化smiles序列d提取出药物的支链,根据smiles的定义规则,即其支链使用“()”括起来,提取出支链,剩余部分为主链;
13.1.2)然后根据字符串匹配提取出主链中常见的子结构;
14.1.3)最后再依据recap(retrosynthetic combinatorial analysis procedure)的逆合成碎片规则将主链进行裂解得到逆合成片段;
15.1.4)整合以上的支链、常见子结构和逆合成片段这3种子结构,作为规范化smiles序列d的子结构集合fd。
16.2)对于靶标氨基酸序列t使用cfm方法提取子结构,cfm方法首先将氨基酸按照化学结构或性质分为8类,通过种类特征映射得到种类序列tc;然后采用不重叠的k-gram序列将tc切割成靶标的功能子结构集合f
t
。
17.步骤c:构建协同特征学习模块,该模块由输入表示和特征学习两部分组成,具体包括:
18.1)在输入表示中,分别对药物的规范化smiles序列d的子结构集合fd和靶标的功能子结构集合f
t
进行初始编码表示,包括:
19.1.1)对药物的规范化smiles序列d的子结构集合fd的表示,首先采用标签编码对药物子结构集合fd进行编码,获得药物的初始表示id;然后将id转化为药物的嵌入表示ed∈r
max_drug_frag_length*embed_size
,其中max_drug_frag_length表示最大的药物子结构集合的大小,embed_size表示嵌入维度;
20.1.2)对靶标的功能子结构集合f
t
进行表示,先用标签编码对靶标子结构集合f
t
进行编码,获得靶标的初始表示i
t
;然后将i
t
转化为其嵌入表示e
t
∈r
max_target_frag_length*embed_size
,其中max_target_frag_length表示最大的靶标子结构集合的大小,embed_size表示嵌入维度。
21.2)在特征学习中,分为药物的特征学习和靶标的特征学习,包括:
22.2.1)在药物特征学习中,将ed作为初始输入,送入卷积神经网络学习药物特征,该卷积神经网络由多个卷积块和最终的池化层组成,每个卷积块由卷积层、激活层指数线性单元(elu)以及批次归一化层(batchnorm)组成,最后采用池化层进行特征降维,得到最终的药物表示vd;
23.2.2)在靶标特征学习中,将e
t
作为初始输入,送入卷积神经网络对靶标特征进行学习,该卷积神经网络由多个卷积块和最终的池化层组成,每个卷积块由卷积层,激活层指数线性单元(elu)以及批次归一化层(batchnorm)组成,最后接入池化层,得到最终的靶标表示v
t
。
24.步骤d:构建预测器,具体包括:
25.1)首先拼接步骤c中最终的药物表示vd和靶标表示v
t
,得到药物-靶标的相互作用表示v;
26.2)然后将v送入多层感知机进行相互作用学习,该多层感知机是一个全连接网络,由多个全连接层和最终的sigmoid激活层组成,除最后一层外,每层全连接后接入一个修正线性单元和丢弃层,防止过拟合;最终得到相互作用预测概率,大于0.5则表示预测两者会发生相互作用,小于0.5则不会。
27.本发明的主要工作和创新包括:针对药物,依据领域知识对药物的规范化smiles序列进行子结构提取,以子结构为基本粒度采用卷积神经网络提取药物的语义特征;针对靶标,对靶标氨基酸序列中的氨基酸根据化学性质进行分类,进行种类映射得到相应的氨基酸种类序列,然后借助自然语言处理方法进行序列分割,以子结构为基本粒度采用卷积神经网络进行语义特征提取;最终拼接药物和蛋白质特征作为相互作用预测器的输入,通过多层全连接网络进行特征学习,从而预测两者是否会发生相互作用。
28.与现有技术相比,本发明的创新性和有效性表现在:提出了一种基于支链挖掘的子结构提取方法bcm(branch chain mining),该方法可以将药物裂解成多种类型的子结构,其中包括支链、常见子结构和逆合成片段,bcm方法首先根据smiles生成规则提取出支链,然后提取出药物里的常见子结构,例如苯环和羟基等,最后使用recap将药物主链裂解成逆合成片段;提出了一种类特征映射方法cfm(cate gory feature mapping),cfm方法将靶标蛋白的氨基酸序列按照氨基酸的化学性质分为8种进行种类映射,然后采用不叠加的k-gram将其切分为子结构;利用卷积神经网络分别学习药物子结构间的协同作用以及靶标子结构间的协同作用;最终进行两者的特征融合作为分类器的输入进行相互作用预测。从实验结果来看,本发明在多个公开数据集上明显优于目前最好的方法,表明该方法能有效去除噪音,提取出药物-靶标相互作用关注的子结构信息,达到较好的效果。
附图说明
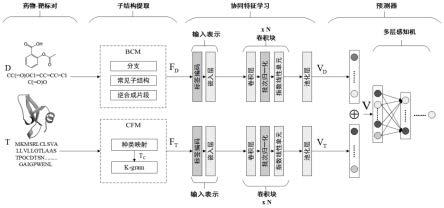
29.图1为本发明的流程示意图;
30.图2是图1中多层感知机的详细流程图。
具体实施方式
31.以下结合附图及实施例对本发明作进一步详细描述。
32.实施例1
33.参阅图1,按下述步骤对本发明进行详细说明:
34.步骤a:药物-靶标对,输入药物的规范化smiles序列d和靶标氨基酸序列t;
35.步骤b:子结构提取,分为药物子结构提取和靶标子结构提取,具体包括:
36.1)药物子结构提取,使用bcm方法提取,具体如下:
37.1.1)首先提取出药物规范化smiles序列d的支链。根据smiles的生成规则,即使用“()”括起来的序列表示支链,通过遍历整个药物规范化smiles序列d,利用栈来匹配每个“(),”提取出“()”内部的子序列即支链,剩余部分连接起来作为主链;
38.1.2)检测出主链中常见的子结构,例如苯环,羟基,醛和羰基。由于这些子结构有
对应的smiles序列,例如苯环可表示为c1=cc=cc=c1,直接通过字符串匹配在主链中去检测是否存在这些子结构;
39.1.3)提取出主链中的逆合成片段。根据recap方法裂解分子,直接借助rdkit工具实现;
40.1.4)将以上3种子结构作为药物规范化smiles序列d的子结构集合fd。
41.通过以上4步可以得到药物的子结构集合,例如阿司匹林的规范化smiles序列d表示为cc(=o)oc1=cc=cc=c1c(=o)o,使用bcm提取出的药物子结构集合为=o(子结构#1),=o(子结构#2),ccoc1=cc=cc=c1co(子结构#3),c1=cc=cc=c1(子结构#4),其中子结构#1和子结构#2是支链,子结构#3是主链,子结构#4是常见的子结构苯环,其中由于主链即子结构#3无法进一步根据recap裂解得到逆合成片段,所以阿司匹林的子结构集合不包含逆合成片段。
42.2)靶标子结构提取,使用cfm方法提取,具体如下:
43.2.1)根据化学性质将氨基酸分为8类,每种类别对应一个类别标签,如表1所示,然后将靶标氨基酸序列t中的氨基酸映射为对应的种类标签,得到种类序列tc;再采用不叠加的k-gram对tc分割成长度为k的子结构,得到子结构集合f
t
。
44.表1
45.[0046][0047]
例如靶标序列t为mkmsrlclsvallvllgtlaastpgcdtsnqakaqrpdfcleppytgpck
……
enl,通过表1进行种类映射后得到种类序列tc为egefgaeafaaaaaaaafaaaffhaecffddagadghcbeachhbfah
……
cda,最后再使用不重叠的k-gram进行序列分割,设置k=3,得到子结构集合f
t
表示为ege fga eaf aaa aaa aaf aaa ffh aec ffd dag adg hcb eac......a。
[0048]
步骤c:协同特征学习,该模块由输入表示和特征学习两部分组成,具体包括:
[0049]
1)在输入表示中,分别对药物的规范化smiles序列d的子结构集合fd和靶标的功能子结构集合f
t
进行初始编码表示,包括:
[0050]
1.1)对药物的规范化smiles序列d的子结构集合fd的表示。首先建立药物子结构字典dd,并按照子结构长度排序,对于一个药物子结构集合fd,采用标签编码进行表示,即每一个子结构利用其在dd中的序号来进行表示,此外由于每个药物产生的子结构数量不同,采用最大的子结构集合大小作为其固定长度,不足则用0填充,获得药物的初始表示id∈r
max_drug_frag_length
其中max_drug_frag_length表示最大的药物子结构集合的大小;然后将id转化为药物的嵌入表示ed∈r
max_drug_frag_length*embed_size
,embedsize表示嵌入维度;
[0051]
1.2)对靶标的功能子结构集合f
t
进行表示。建立靶标功能子结构字典d
t
,并按照子结构长度排序,对于一个靶标子结构集合f
t
,采用标签编码进行表示,即每一个子结构利用其在d
t
中的序号来进行表示,此外由于每个靶标产生的子结构数量不同,采用最大的子结构集合大小作为其固定长度,不足则用0填充,获得靶标的初始表示i
t
∈r
max-target_fragg_length
,其中max_target_frag_length表示最大的靶标子结构集合的大小;然后将it转化为其嵌入表示e
t
∈r
max_target_frag_length*embed_size
,embed_size表示嵌入维度。
[0052]
2)在特征学习中,分为药物特征学习和靶标特征学习,包括:
[0053]
2.1)在药物特征学习中,将ed作为初始输入,送入卷积神经网络学习药物特征,该
卷积神经网络由多个卷积块和最终的池化层组成,每个卷积块由卷积层、激活层指数线性单元(elu)以及批次归一化层(batchnorm)组成,最后采用池化层进行特征降维,得到最终的药物表示vd,具体如下:
[0054]
2.1.1)卷积层,表示药物嵌入ed经过一维卷积操作,得到和原来相同维度的特征矩阵,卷积操作可表示为公式2,其中ed表示药物嵌入表示,f表示卷积操作,wj和bj分别表示第j个卷积层的权重矩阵和偏置;
[0055]
ed∈r
max_drug_frag_length*embed_size
ꢀꢀꢀ
(1)
[0056]fj
=f(ed*wj+bj)
ꢀꢀꢀ
(2)
[0057]
2.1.2)批次归一化,对fj进行归一化,其计算如下所示,其中是的批次集合,m表示批次集合的大小,xs表示中第s个值,和是批次内的平均值和方差,∈是偏置,是xs经过标准化后的值,γ和β是学习参数,最终的输出为ys;
[0058][0059][0060][0061]
2.1.3)经由归一化函数后,选择elu作为激活函数,可表示为公式(6),其中xi表示第i个神经元的值,α是参数;
[0062][0063]
2.1.4)经由n个卷积块后,接入池化层,降低特征图维度,提高网络训练速度。
[0064]
2.2)在靶标特征学习中,将e
t
作为靶标特征初始输入,送入卷积神经网络对靶标特征进行学习,该卷积神经网络由n个卷积块和最终的池化层组成,每个卷积块由卷积层、激活层指数线性单元(elu)以及批次归一化层(batchnorm)组成,最后接入池化层,得到最终的靶标表示v
t
。
[0065]
步骤d:送入预测器,预测药物和靶标两者是否会发生相互作用,具体包括:
[0066]
1)首先,拼接步骤c中最终的药物表示vd和靶标表示v
t
得到药物-靶标的相互作用表示v;
[0067]
2)然后将v送入多层感知机进行相互作用学习,多层感知机是一个全连接网络,如图2所示,该网络由多个全连接层和最终的sigmoid激活层组成,除最后一层外,每层全连接后接入一个修正线性单元和丢弃层,防止过拟合;最终得到相互作用概率,大于0.5则表示预测两者会发生相互作用,小于0.5则不会。
[0068]
在本实施例中,通过adam优化器对网络参数进行优化,使用二元交叉熵作为损失函数,可表示为:
[0069][0070]
其中,m是参与训练的样本数,pi是样本i预测出的概率,yi表示样本i的真实标签。
[0071]
实施例2
[0072]
在本实施例中,使用pytorch实现本发明,并在四个公开的数据集上进行实验,分别为c.elegans、human、davis和bindingdb数据集,数据分布如表2所示。
[0073]
表2
[0074][0075]
1)c.elegans和human数据集包含了最详细的已知和临床激酶抑制剂在整个激酶组的反应性,并且完全覆盖了人类蛋白质激酶组,其正样本来自于置信度最高的生化数据库drugbank数据库和matador数据库,包含1717种药物和1857种靶标,且正负样本平衡;
[0076]
2)davis m i,hunt j p,herrgard s等人通过将各种资源整合到一个系统的筛选框架中,并受“相似的化合物可能与与相应已知目标蛋白相似的蛋白质发生相互作用”启发,假设与化合物的每个已知/预测靶标不同的蛋白质不太可能被该化合物靶向,另一方面,与靶向蛋白质的任何已知/预测化合物不相似的化合物也不多可能靶向这种蛋白质,然后筛选出高度可信的负样本,构建了davis数据集,包含64种药物,379种靶标,正负样本严重失衡;
[0077]
3)bindingdb数据集来自bindingdb数据库,其数据是从文献研究中提出的,侧重于收集作为药物靶标或者候选靶标的蛋白质信息,该数据集包含7134种药物,1251种靶标,正负样本失衡。
[0078]
在实验中,将卷积块数n设为2,优化器学习率设为1e-5,批次为32,允许最多运行100个迭代,并设置早停止,为防止模型过拟合,设置丢失率为0.5;在实验评估中,使用auc、precision和recall作为衡量模型性能的指标,本发明以8:1:1的比例划分为训练集,验证集和测试集,选择验证集中最优auc的模型作为最佳的模型,然后在测试集上进行对比评估。
[0079]
表3
[0080][0081]
表4
[0082][0083]
表5
[0084][0085]
表6
[0086][0087]
本发明提供了一种基于生物子结构去预测药物-靶标相互作用的深度学习方法,并在4个公开的数据集上进行实验,与其他机器学习方法进行性能比较。表3和表4分别是本发明在c.elegans和human这两个正负样本均衡数据集上的对比实验结果,表5和表6分别是本发明在davis和bindingdb这两个正负样本分布不均衡数据集上的实验结果,从以上结果可以看出本发明无论在数据集分布均衡或不均衡的情况下,其预测都能取得更好的效果,说明了本发明基于子结构的思想去对药物-靶标相互作用预测任务建模是切实有效的,可以显式增强药物和靶标的药理特征,也体现了本发明的通用性;此外与浅层机器学习方法或深度学习方法相比,其预测性能都能取得领先的效果,说明了本发明采用卷积神经网络挖掘药物和靶标子结构间的协同作用,对预测相互作用具有积极意义。
[0088]
以上各实施例只是进一步说明本发明,并非用以限制本发明,凡为本发明等效实施,均应包含于本发明的权利要求范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1