评价葡萄糖激酶激活剂治疗糖尿病疗效和安全性的方法与流程
1.本发明涉及评价葡萄糖激酶激活剂治疗糖尿病患者的疗效和安全性的方法。
背景技术:
2.糖尿病由于其增加的流行率及相关的健康风险而成为主要的公共健康问题。糖尿病主要分为1型糖尿病(胰岛素依赖性糖尿病,t1md)和2型糖尿病(非胰岛素依赖性糖尿病,t2md)。t2md患者的典型临床症状是高血糖,是由胰岛素耐受性和胰岛素分泌受损引起的,高血糖会引起相关的发血管和微血管的并发症,控制血糖水平是治疗该病症的主要手段,目前二甲双胍作为t2md的治疗药物已经广泛应用,并且有多种其他降糖药物可选择,但是在服药的患者中仅有一部分患者能够达到理想血糖状态,且由于β细胞功能的进行性下降,继发性失败在服用上述药物的患者中很常见。此外,上述药物都存在一定的副作用和风险,一定程度上会影响患者的生活质量,还会影响治疗的选择和效果。
3.已糖激酶(hk)是人体葡萄糖代谢途径中的第一个限速酶,葡萄糖激酶(gk)作为hk家族中的一员,在维持血糖稳定方面具有至关重要的生理作用,当体内血糖升高并超过正常范围时gk被激活,通过增加胰岛素释放和促进肝脏葡萄糖的利用双重作用机制来降低血糖,当血糖降低时,gk活性则随之降低。葡糖激酶激活剂(glucokinase activator,gka),能够在血糖浓度升高的情况下激活gk,以葡萄糖依赖性方式增加葡萄糖刺激的胰岛素分泌、提高肝脏葡萄糖摄取以及减少肝脏葡萄糖输出,从而调节血糖稳态。到目前为止,已经开发了多个gka,然而,目前仅在全球范围内研究了四种候选药物(例如ttp399、dorzagliatin、globalagliatin和pb-201),大多数葡萄糖激酶激活剂没有进入后期开发阶段,存在着随着时间的推移疗效丧失和安全问题,如出现低血糖、高脂血症和肝功能损害等不良作用。因此,设计和开发一种能够针对葡萄糖激酶激活剂评价其在治疗糖尿病患者中的疗效和安全性的方法是亟需解决的问题。
4.pb-201为一种新型葡萄糖激酶部分激活剂(gka),同时作用于肝脏和胰腺。相较于肝脏选择性gka,疗效更佳,预期不会因肝细胞核内葡萄糖激酶的过度耗竭而出现疗效降低的情况,预期也不会导致转氨酶及甘油三酯的明显增高。pb-201作为部分激动剂,与葡萄糖激酶的结合和解离较快,药效呈现瞬时启动/消失的特点,对低血糖的敏感性好,因而起效缓和,效能适中,且低血糖风险低。pb-201在境外已完成非临床及七项i期和两项ii期临床研究,共有565例t2dm受试者至少接受一次pb-201给药,结果显示pb-201能够降低t2dm患者的hba1c水平,且在研究的剂量范围内具有较好的安全耐受性。基于pb-201已完成的临床研究,本发明旨在提供一种方法能够确定最佳的pb-201剂量,以用于未来的验证性试验,并评估pb-201在未接受药物治疗的t2dm中国患者中的以明确pb-201在中国2型糖尿病人群中的安全性、耐受性、药代动力学和药效动力学特征,并选择出合适的剂量进行评价,明确药物的量效关系。
技术实现要素:
5.(一)解决的技术问题
6.针对现有技术的不足,本发明提供了评价葡萄糖激酶激活剂治疗糖尿病患者的疗效和安全性的方法,所述方法采用随机、双盲、安慰剂对照、四交叉、四周期自身对照设计方法,并利用持续性血糖检测(cgm)技术,该方法能够明确葡萄糖激酶激活剂在中国2型糖尿病人群中的安全性、耐受性、药代动力学和药效动力学特征,并选择出合适的剂量进行评价,明确药物的量效关系。
7.(二)技术方案
8.为实现以上目的,本发明通过以下技术方案予以实现:
9.本发明提供了评价葡糖糖激酶激活剂治疗糖尿病患者的疗效和安全性的方法,所述方法采用随机、双盲、安慰剂对照、四交叉、四周期自身对照设计方法,并利用持续性血糖检测(cgm)技术。
10.所述方法具体按照以下步骤:
11.(1)筛选符合试验标准的受试者16例;
12.(2)16例受试者按照1:1:1:1随机分配成4组,每组4例受试者,分别为1-4组,每组受试者分别按照安慰剂、研究药物剂量1、研究药物剂量2、研究药物剂量3共4种剂量方案接受4种顺序治疗,具体治疗顺序周期和药物剂量如表1所示;
13.表1 1-4组受试者治疗顺序及药物剂量
[0014][0015]
(3)受试者按照上述剂量方案每天早午餐前30分钟服用研究药物,温水吞服,每个治疗周期给药7天,每个治疗周期间隔7-14天的洗脱期;
[0016]
(4)安全性评价:研究期间对葡萄糖激酶激活剂的安全耐受性进行观察统计与分析;
[0017]
(5)药代动力学评价:利用hplc-ms/ms方法对服用pb-201片剂后患者血浆中葡萄糖激酶激活剂及其代谢产物的浓度进行检测,采用phoenix winnonlin软件的非房室分析方法(linear up log dowm)计算葡萄糖激酶激活剂及其代谢产物的pk参数,并进行描述性统计;
[0018]
(6)药效学评价:按剂量和天数对空腹及餐后2小时静脉血糖、血清c肽和血清胰岛
素浓度进行描述性统计;
[0019]
(7)动态血糖检测:按照研究药物剂量1、研究药物剂量2、研究药物剂量3,三个研究药物剂量组和安慰剂组分别计算以下指标的平均值:指定观测日葡萄糖平均值、指定观测日葡萄糖在目标范围(血糖3.90-10.0mmol/l)内的时间百分比,指定观测日葡萄糖低于目标范围的时间百分比、指定观测日葡萄糖高于目标范围的时间百分比、指定观测日预估糖化血红蛋白;
[0020]
(8)对步骤(4)至步骤(7)数据进行统计、处理、分析、得出结论。
[0021]
上述方法中所述葡萄糖激酶激活剂包括但不限于ttp399、dorzagliatin、globalagliatin或pb-201。
[0022]
按照上述方法能够明确葡萄糖激酶激活剂pb-201在中国2型糖尿病人群中的安全性、耐受性、药代动力学和药效动力学特征,并选择出合适的剂量进行评价,明确药物的量效关系,葡萄糖激酶激活剂pb-201均可显著降低血糖并呈剂量依赖性。
[0023]
按照上述方法能够证明葡萄糖激酶激活剂pb-201在中国人群中能有效降低空腹及餐后血糖,并且安全性、耐受性良好,极大降低患者在给药后出现低血糖的风险,显著增加血糖在目标范围内时间(tir),并确定了100+100mg pb-201为后续研究的最佳剂量。
[0024]
上述方法中,步骤(2)所述研究药物剂量1-3是基于已有的针对葡萄糖激酶激活剂的临床研究数据确定的合适的剂量范围;其中葡萄糖激酶激活剂pb-201的剂量设置分别为50+50mg pb-201、100+50mg pb-201、100+100mg pb-201。
[0025]
(三)有益效果
[0026]
本发明提供了评价葡萄糖激酶激活剂治疗t2dm患者的疗效和安全性的方法。本发明创新性的设计了随机、双盲、安慰剂对照、四交叉、四周期自身对照方法,摒弃了在生物等效性试验中最主要使用的“2
×
2交叉”方法,以每个受试者作为自身对照,有效避免个体间差异带来的偏差,明显减少样本数量,增加了研究结果的精确性、可靠性,能够更好地评价葡萄糖激酶激活剂的剂量-暴露-效应之间的关系,同时在国内率先使用了持续性血糖检测(cgm)技术。利用本发明所述方法证明了葡萄糖激酶激活剂pb-201在中国人群中能有效降低空腹及餐后血糖,并且安全性、耐受性良好,极大降低患者在给药后出现低血糖的风险,显著增加血糖在目标范围内时间(tir)。利用本发明所述方法能够明确葡萄糖激酶激活剂pb-201在中国2型糖尿病人群中的安全性、耐受性、药代动力学和药效动力学特征,并选择出合适的剂量进行评价,明确药物的量效关系,发现pb-201均可显著降低血糖并呈剂量依赖性,同时明确了100+100mg pb-201为更合适的临床ⅲ期治疗剂量。利用本发明所述方法得到的相关数据,获得了国家药审评中心(cde)对pb-201豁免ⅱ期临床研究直接进入ⅲ期临床研究。
附图说明
[0027]
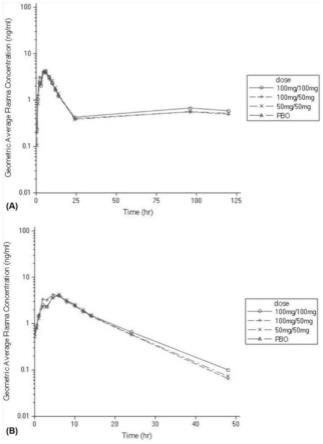
图1-a和图1-b分别为t2dm患者单次口服pb-201(早午餐前服药)后即给药第1天和连续给药第7天后的不同剂量下pb-201的血药浓度-时间曲线图。
[0028]
图2-a和图2-b分别为t2dm患者单次口服pb-201(早午餐前服药)后即给药第1天和连续给药第7天后的不同剂量下wi-0800的血药浓度-时间曲线图。
[0029]
图3为pb-201和wi-0800剂量比例的线性回归分析数据图。
[0030]
图4-a为通过cgm测量的服用安慰剂和不同剂量pb-201的患者的24小时血糖动态曲线图,其中实线代表血糖的平均值,虚线代表第一和第三分位数;图4-b为安慰剂组和三个剂量pb-201组在连续24小时动态血糖检测过程中血糖浓度在超出范围时间(tar)、低于范围时间(tbr)及在范围内时间(tir)的时间百分比。
具体实施方式
[0031]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0032]
评价葡萄糖激酶激活剂治疗t2dm患者的疗效和安全性的方法
[0033]
1筛选符合试验标准的受试者16例
[0034]
2试验设计
[0035]
研究药物:pb-201,片剂,规格50mg、100mg;安慰剂(pb-201模拟片),规格50mg、100mg。
[0036]
16例受试者按照1:1:1:1随机分配成4组,每组4例受试者,分别为1-4组,每组受试者分别按照安慰剂、50+50mg pb-201、100+50mg pb-201、100+100mg pb-201剂量方案接受4种顺序治疗,具体治疗顺序周期和药物剂量如表1所示;
[0037]
表1 1-4组受试者治疗顺序及药物剂量
[0038][0039][0040]
给药方法:
[0041]
受试者按照上述剂量方案每天早餐前30分钟服用pb-201或安慰剂,温水吞服,早餐给药后约4小时进午餐,并在进午餐前30分钟服用pb-201或安慰剂;每个治疗周期给药7天,每周期第1、7天,要求所有受试者在给药后4小时内避免躺卧(除生命体征和心电图测量所需外)和进食(除研究需要外),给药后2小时内禁止饮水;每个治疗周期间隔7-14天的洗脱期。
[0042]
3评价指标
[0043]
3.1安全性评价
[0044]
研究期间安全耐受性观察指标如下:
[0045]
(1)不良事件/严重不良事件(包括低血糖事件);
[0046]
(2)实验室检查(血常规、血生化、尿常规);
[0047]
(3)生命体征(体温、心率、呼吸、血压);
[0048]
(4)体格检查(包括头部、五官、颈部、胸廓、背部、心脏、肺部、腹部、淋巴结、皮肤、四肢、神经肌肉系统);
[0049]
(5)12导联心电图检查(包括pr间期、qrs间期、qtc间期)。
[0050]
3.2药代动力学(pk)
[0051]
利用hplc-ms/ms方法对服用pb-201片剂后患者血浆中pb-201何其代谢产物(wi-0800)的浓度进行检测,采用phoenix winnonlin软件的非房室分析方法(linear up log dowm)计算pb-201及其代谢产物(wi-0800)的下列pk参数:
[0052]
(1)每个周期的第1天:c
max
、达峰时间(t
max
)、auc
0-24
、auc
inf
、auc
last
、清除率(cl/f)、t
1/2
t
1/2
、表现分布容积(vz/f)、auc
_%extrap
、消除速率常数(kel);
[0053]
(2)每个周期的第7天:t
max
、auc
0-24ss
、给药前浓度(c
trough
)、0~24小时内的平均浓度(c
avss
)、c
minss
、c
maxss
、清除率(cl/f
ss
)、t
1/2ss
、表现分布容积(vz/f
ss
)、以auc
0-24ss
和auc
0-24
算的稳态蓄积比(r
auc1
)、以c
max
和cmaxss计算的稳态蓄积比(r
cmax
)、以auc
0-24ss
和auc
inf
计算的稳态蓄积比(r
auc2
)、auc
infss
、auc
lastss
、auc
_%extrapss
、消除速率常数(kel
ss
)、波动系数(ptf)。
[0054]
3.3药效学(pd)
[0055]
观察指标如下:
[0056]
(1)每个治疗周期的第7天,每个pb-201剂量水平与安慰剂相比,空腹静脉血糖相对于时间匹配基线的变化;
[0057]
(2)每个治疗周期的第7天,每个pb-201剂量水平与安慰剂相比,标准餐下餐后2小时静脉血糖相对于时间匹配基线(每个周期的第0天)的变化;
[0058]
(3)每个治疗周期的第7天,每个pb-201剂量水平与安慰剂相比,空腹血清胰岛素和c肽相对于时间匹配基线(每个周期的第0天)的变化;
[0059]
(4)每个治疗周期的第7天,每个pb-201剂量水平与安慰剂相比,标准餐下餐后2小时血清胰岛素和c肽相对于时间匹配基线(每个周期的第0天)的变化;
[0060]
(5)每个治疗周期,每个pb-201剂量水平与安慰剂相比,动态许堂监测相关指标(如平均血糖水平、日内/日间血糖波动等)的变化,以及治疗后hbalc计算值相对于基线(每个周期的第0天的变化)。
[0061]
4数据分析
[0062]
4.1安全性分析
[0063]
依照药事管理的标准医学学术预计对ae进行编码,包括严重不良事件(sae)在内的所有ae都根据系统器官分类和首选术语列表。应分周期和剂量组对ae进行统计,报告ae、ae受试者的例数、例次和百分比,按照meddra系统器官分类和首选术语列表,并按照与研究用药物的关系和严重程度汇总ae并列表。对死亡、sae和导致退出研究的ae进行汇总并列表。
[0064]
按照不同阶段对受试者的12导联ecg结果进行汇总,列出服药前后心电图的变化情况。按照受试者编号、组别对心电图异常结果进行列表。
[0065]
按照不同阶段对计划评价时间点中的受试者的生命体征进行汇总,描述用药前后生命体征的变化。列出例数(n)、算数平均值(mean)、标准差(sd)、最小值(min)、中位数(median)和最大值(max),并列出异常结果清单。
[0066]
对每次评价的体格检查数据进行列表。对异常结果按受试者列表汇总。
[0067]
按照给药阶段对研究期间的合并用药进行列表。
[0068]
4.2药代动力学分析
[0069]
按给药剂量(50+50mg、100+50mg、100+100mg)进行分组,分别对不同剂量组的受试者的pb201及wi-0800的血药浓度进行统计描述。按不同给药剂量,分别绘制个体受试者的血浆浓度-时间曲线。
[0070]
采用phoenix winnonlin软件的非房室分析方法(linear up log dowm)计算pb-201及其代谢产物(wi-0800)的pk参数,并进行描述性统计。
[0071]
使用power模型分析pb-201及wi-0800的暴露auc、c
max
与剂量之间的线性关系。
[0072]
4.3药效动力学分析
[0073]
空腹及餐后2小时静脉血糖、c肽和胰岛素:
[0074]
按剂量和天数对空腹及餐后2小时静脉血糖、血清c肽和血清胰岛素进行描述性统计。按剂量组第7天药效学参数减去基线期(第0天)对应的药效学参数得到药效学参数相对基线变化值;pb-201的3个剂量组药效学参数相对基线变化值减去安慰剂组药效学参数相对基线变化值(扣除安慰剂)再与安慰剂组药效学参数相对基线变化值进行最小二乘均数(lsm)和90%置信区间(ci)(用mmol/l表示)的统计分析比较差异,其结果经假设检验,当p小于0.05时,有显著差异。
[0075]
4.4动态血糖检测
[0076]
计算个体的指定观测日葡萄糖平均值、指定观测日葡萄糖在目标范围(血糖3.90-10.0mmol/l)内的时间百分比、指定观测日葡萄糖低于目标范围的时间百分比、指定观测日葡萄糖高于目标范围的时间百分比、指定观测日预估糖化血红蛋白(其中个体的给药日预估糖化血红蛋白方程为:a1c
%
=(avgsg
mg/dl
+46.7)/28.7,a1c
%
=(avgsg
mmol/l
+2.59)/1.59,其中a1c
%
为预估的糖化血红蛋白水平,avgsg
mg/dl
是单位以mg/dl表示的平均血糖,avgsg
mm
ol
/l
是单位以mmol/l表示的平均血糖),以及4个剂量组上述指标的平均值。
[0077]
按照50+50mg、100+50mg、100+100mg剂量组和安慰剂组分别计算以下指标的平均值:指定观测日葡萄糖平均值、指定观测日葡萄糖在目标范围(血糖3.90-10.0mmol/l)内的时间百分比,指定观测日葡萄糖低于目标范围的时间百分比、指定观测日葡萄糖高于目标范围的时间百分比、指定观测日预估糖化血红蛋白。
[0078]
5结果
[0079]
5.1安全性结果
[0080]
本研究共15例受试者发生了34例次不良事件,严重程度均为轻度。共有2例受试者发生了2例次与研究药物相关的不良事件,均为相对低血糖,均发生在50/50mg剂量组。5例受试者因不良事件接受合并用药。研究期间未发生严重不良事件或导致受试者死亡的不良事件。仅1例受试者因达到符合方案规定的高血糖退出标准退出研究。本研究未发现被研究
者判定为与研究药物相关的且有临床意义的安全性实验室指标、心电图、生命体征和体格检查的异常或变化。
[0081]
5.2药代动力学结果
[0082]
在50+50mg、100+50mg和100+100mg剂量组,中国t2dm患者单次口服pb-201(早午餐前服药)后,pb-201迅速吸收,血药浓度分别在5.25(4.50,8.00)h、4.50(2.00,6.00)h和6.00(4.50,6.00)h达到峰浓度;c
max
分别为483
±
118ng/ml、660
±
144ng/ml和929
±
210ng/ml(如图1-a所示),随后pb-201血药浓度呈双指数曲线下降;药物在体内分布广泛,vz/f分别为165
±
51.6l、191
±
64.4l和203
±
73.3l;清除率较低(与肝或肾血流量比较),cl/f分别为21.8
±
6.18l/h、21.6
±
4.81和21.8
±
5.74l/h;半衰期t
1/2
分别为5.72
±
1.91h、6.11
±
1.37h和6.41
±
1.33h。
[0083]
pb-201每日早午给药2次,连续给药4日(96h)后,3个剂量组(50+50mg、100+50mg和100+100mg)的pb-201血浆药物浓度均达到稳态。连续给药7天,第7天,3个剂量组pb-201血药浓度分别在6.00(4.50,8.00)h、4.50(2.00,6.00)h和6.00(4.50,6.00)h达到峰浓度(如图1-b所示),c
maxss
分别为462
±
82.5ng/ml、710
±
139ng/ml和896
±
190ng/ml,消除相半衰期t
1/2ss
分别为8.01
±
2.54h、8.26
±
2.42h和9.55
±
2.83h;r
auc1
分别为1.07
±
0.237、1.12
±
0.123和1.10
±
0.186,r
auc2
分别为0.990
±
0.239、1.03
±
0.113和1.00
±
0.164,r
cmax
分别为1.01
±
0.292、1.10
±
0.231和0.988
±
0.193,结果提示pb-201在50/50-100/100mg剂量范围内连续早午餐给药7天后pb-201在体内没有蓄积。
[0084]
在50+50mg、100+50mg和100+100mg剂量组,中国t2dm患者第1天口服pb-201(早午餐前服药)后,代谢物wi-0800分别在10.0(10.0,12.0)h、10.0(6.00,14.0)h和10.0(8.00,12.0)h达到峰浓度(如图2-a所示),c
max
分别为474
±
111ng/ml、699
±
187ng/ml和919
±
245ng/ml,随后呈双指数曲线下降,代谢物wi-0800在患者体内的暴露量高于原型药pb-201,mrauc
0-24
分别为1.78
±
0.332、1.85
±
0.378和1.80
±
0.372。pb-201每日早午餐前服药,连续给药4日(96h)后,3个剂量组的wi-0800药物浓度基本达到稳态,其中100/100mg剂量组wi-0800浓度在144h的浓度相较于96h的浓度有约10.2%的升高。在给药第7天,各剂量组代谢物wi-0800的c
maxss
分别为717
±
167ng/ml、1094
±
237ng/ml和1378
±
258ng/ml(如图2-b所示),t
maxss
分别为10.0(6.00,12.0)h、10.0(6.00,10.0)h和10.0(0.00,12.0)h,r
auc1
分别为1.73
±
0.277、1.78
±
0.175和1.78
±
0.296,r
cmax
分别为1.53
±
0.270、1.60
±
0.213和1.56
±
0.337,结果提示连续给药7天后代谢物wi-0800在体内暴露增加。在序惯的四个研究周期内,给药第1天和第7天wi-0800和pb-201暴露量(c
max
、auc
last
、auc
0-24
)均值的比值未见增加趋势,在50/50mg、100/50mg和100/100mg剂量范围内,wi-0800和pb-201的暴露量均值的比值未见增加趋势。
[0085]
在50+50mg、100+50mg和100+100mg剂量组,受试者口服pb-201后,第1天的pb-201的c
max
、auc
last
、auc
inf
和第7天c
maxss
的点估计值均高于1,均落在判断区间内,90%ci上限超出判断区间,第7天的pb-201的auc
lastss
、auc
infss
的点估计值和90%ci均落在判断区间内。结果提示pb-201单次给药后呈近似线性,早午餐前给药,连续给药达稳态后,auc
ss
呈现剂量线性特征,c
maxss
呈现近似剂量线性特征。wi-0800第1天pk参数(c
max
、auc
last
)和第7天pk参数(c
maxss
、auc
lastss
)的点估计值均高于1,均落在判断区间内,90%ci上限超出判断区间,结果提示pb-201代谢物wi-0800暴露量在50/50~100/100mg pb-201剂量范围内,呈现近似线性
特征,暴露量增加略超过剂量增加比例。结果如图3所示。
[0086]
5.3药效动力学结果
[0087]
(1)安慰剂组第7天空腹血糖与基线相比lsm(90%ci)升高0.307(-0.161,0.775)mmol/l;各剂量组相对基线和相对安慰剂的变化值及统计学差异检验结果请见下表2。
[0088]
表2各剂量组第7天空腹血糖相对基线和相对安慰剂的变化值
[0089][0090][0091]
(2)安慰剂组第7天餐后2小时血糖相对基线升高0.827(0.195,1.459)mmol/l;各剂量组相对基线和相对安慰剂的变化值及统计学差异检验结果请见下表3。
[0092]
表3各剂量组第7天餐后2小时血糖相对基线和相对安慰剂的变化值
[0093][0094]
(3)不同剂量组的空腹及餐后2小时c肽、胰岛素,第7天相对于基线的变化与安慰剂组无显著差异,p值》0.05;
[0095]
(4)安慰剂组、50+50mg、100+50mg和100+100mg组动态血糖在目标范围(3.90~10.0mmol/l)的时间百分比分别为49.380%、49.210%、56.130%和63.330%;葡萄糖平均值分别为10.680mmol/l、10.820mmol/l、10.020mmol/l和8.700mmol/l;hba1c基线值均值分
别为8.931%、8.979%、8.993%、8.993%,预估hba1c分别为8.350%、8.435%、7.930%和7.105%,与基线值差值的均值分别为-0.581%、-0.544%、-1.063%、-1.888%。
[0096]
5.4动态血糖检测结果
[0097]
根据cgm检测结果可知,与安慰剂相比,随着pb-201剂量的增加,每日血糖动态的轨迹较低,如图4-a所示,平均血糖为安慰剂10.680
±
1.990mmol/l和pb-201三种剂量水平分别为10.820
±
2.670mmol/l、10.020
±
2.010mmol/l、8.700
±
2.140mmol/l。范围内时间(tir)随着pb-201剂量的增加而增加,其中100+50mg、100+100mg剂量组的tir高于安慰剂组(安慰剂组和50+50mg、100+50mg、100+100mg三个剂量组的tir分别为49.380%、49.210%、56.130%、63.330%,如图4-b所示)。安慰剂组和三个剂量水平的pb-201分别只有0.310%、0%、0.070%、4.600%的时间处于低范围的时间(tbr)(指血糖浓度<3.9mmol/l),该结果表明安慰剂和pb-201的低血糖发生率较低。安慰剂和三种剂量水平的pb-201的基线hba1c分别为8.931%、8.979%、8.993%和8.993%,治疗后平均估计安慰剂和三种剂量水平的pb-201的hba1c分别为8.350%(68mmol/mol)、8.435%(69mmol/mol)、7.930%(63mmol/mol)和7.105%(54mmol/mol)。与安慰剂基线相比,接受100+50mg和100+100mg pb-201治疗的患者的hba1c估计值的降低呈剂量依赖性且数值更大(安慰剂和三种剂量水平的pb-201的hba1c估计值分别为-0
·
581
±
1.200%或-6.44
±
13.1mmol/mol、-0
·
5445
±
1.654%或-5.9
±
18.1mmol/mol、-1.063
±
1.236%或-11.6
±
13.5mmol/mol、-1
·
888
±
1.381%或-20.6
±
15.1mmol/mol)
[0098]
6结论
[0099]
中国未经药物治疗的成人t2dm受试者每日早、午口服50+50mg、100+50mg和100+100mg的pb-201片剂,连续给药7天后,安全性和耐受性良好。所有不良事件均为轻度,无严重不良事件发生,无严重低血糖发生。
[0100]
50+50mg、100+50mg和100+100mg的pb-201连续给药7天,4天(96h)后谷浓度达到稳态,血浆浓度在4.50h至6.00h达峰,平均t
1/2ss
在8.01h至9.55h,pb-201在体内没有蓄积。
[0101]
受试者第1天及第7天口服50+50mg、100+50mg和100+100mg pb-201后,代谢物wi-0800血药浓度均在10h左右达峰;代谢物暴露量高于原药pb-201,给药后第1天mrauc
0-24
在1.78至1.85。连续给药4天后谷浓度基本达到稳态。
[0102]
在50+50mg、100+50mg和100+100mg剂量范围内,pb-201早午餐前连续给药达稳态后,auc
ss
呈现剂量线性特征,c
maxss
呈现近似剂量线性特征。pb-201代谢物wi-0800暴露量在该剂量范围内,呈现近似线性特征,暴露量增加略超过剂量增加比例。
[0103]
连续7天给药后,50+50mg剂量组空腹血糖与安慰剂组相比无统计学差异,100+50mg和100+100mg剂量组空腹血糖相比于安慰剂组显著降低,呈现剂量相关性趋势。50+50mg、100+50mg和100+100mg剂量组餐后2小时血糖,相比于安慰剂组,均有显著降低,呈现剂量相关性趋势;50+50mg、100+50mg和100+100mg剂量组动态血糖指标的变化呈剂量相关性趋势;不同剂量组的受试者空腹及餐后2小时c肽、胰岛素无显著差异。
[0104]
根据上述结果,能够证明将pb-201的剂量增加到100mg,每天给药两次,对患者具备良好的安全性和耐受性,更能够有效地控制血糖;明确了100+100mg pb-201为更合适的临床ⅲ期治疗剂量;成功免除常规大样本量临床ⅱ期的量效关系探索研究。
[0105]
以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例
对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1