一种基于多样本全外显子测序的变异过滤方法与流程
1.本发明属于二代测序变异分析技术领域,具体涉及一种基于多样本全外显子测序的变异过滤方法。
背景技术:
2.随着高通量测序技术的发展,单张芯片单次上机测序可产出的碱基数最高可超过1t,按单个全外显子测序样本10g个碱基计,可容纳超过100个样本。全外显子测序技术(whole exome sequencing,以下简称wes)是指基于人类基因组外显子区dna捕获富集后进行高通量测序的技术。
3.wes原始序列经过序列预处理、序列比对、变异识别、信息注释和变异过滤的生物信息学分析,并由遗传分析人员综合考虑人群频率、变异质量、变异对蛋白的影响程度、表型关联和病例报道等信息,从数百万个变异中找到引起相应疾病的1-2个变异位点,以上是目前常见的wes数据分析方案。
4.现有针对wes变异位点的主要过滤方法包括以下步骤:1)下载gnomad数据库,构建符合注释软件要求的人群频率文件;2)下载clinvar数据库,构建符合注释软件要求的变异致病性文件;3)采用变异注释软件,如vep(ensembl variant effect predictor),对变异识别输出的多个vcf文件分别进行数据库注释,获得每个变异位点的人群频率、致病性信息;4)根据acmg指南,构建质量和人群频率阈值、致病性规则,过滤掉低质量、高频率和良性变异,得到可用于医学遗传分析的变异结果文件。
5.综上,现有变异过滤方案存在的问题如下:
6.(1)变异注释是一个非常消耗计算机资源和时间的过程,采用变异注释软件对每个样本的vcf文件分别进行人群频率、致病性信息进行注释,会导致样本间相同的变异位点被重复多次注释,造成了大量的计算机资源和时间的浪费;
7.(2)二代测序技术具有测序批次间的质量差异,基于单个样本的过滤方案,对于单个批次内因为文库构建、探针捕获或测序环节造成的特定基因组区域的假阳性位点,很难被过滤;
8.(3)过滤环节未设置白名单,会导致漏掉可能致病的变异。
技术实现要素:
9.针对现有技术存在的问题,本发明提供了一种基于多样本全外显子测序的变异过滤方法。本发明通过构建变异位点黑名单和白名单,结合待分析样本的变异质量和变异检出频率,对多个样本的变异位点进行联合过滤。
10.本发明首先提供了一种基于多样本全外显子测序的变异过滤方法,其包括如下步骤:
11.1)挑选变异分类数据库clinvar中clinsig字段为pathogenic,likely_pathogenic和uncertain_significance的变异,作为白名单;
12.挑选数据库gnomad中人群频率大于0.02的变异,与clinvar数据库中clnrevstat字段为practice_guideline/reviewed_by_expert_panel,且clinsig字段为benign/likely_benign的变异取并集后,与白名单取差集,从而构建变异黑名单;
13.2)选取质量阈值和频率阈值,过滤低质量和高频变异;
14.3)根据黑名单进行变异过滤;
15.4)对步骤3)所得结果进行信息注释;
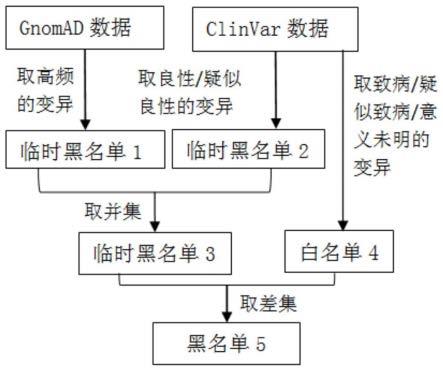
16.5)对步骤4)所得结果按样本进行拆分,得到每个样本注释后的结果。
17.作为本发明的优选方案,所述的步骤1)具体包括如下步骤:
18.1.1下载gnomad数据库提供的wes的vcf文件,提取其中filter字段为pass,且人群频率大于0.02的变异位点,生成“临时黑名单1”;
19.1.2下载clinvar数据库提供的vcf文件,提取clnrevstat字段为practice_guideline或reviewed_by_expert_panel,且clinsig字段为benign或likely_benign的位点,生成“临时黑名单2”;
20.1.3对“临时黑名单1”和“临时黑名单2”中的位点取并集,生成“临时黑名单3”;
21.1.4提取clinvar数据库中clinsig字段为pathogenic、likely_pathogenic和uncertain_significance的位点生成“白名单4”;
22.1.5将“临时黑名单3”与“白名单4”取差集,生成vcf格式的“黑名单5”。
23.作为本发明的优选方案,步骤2)具体为:
24.对多样本的wes联合识别变异数据进行分析,变异位点若符合以下任一条件,则删除,输出最终保留的结果为“过滤结果1”:
25.a.每个样本的测序覆盖深度dp的最大值《10;
26.b.等位总数an》12且变异频率af》0.5;
27.c.等位总数an》30且变异频率af》0.2;
28.d.等位总数an》100且变异频率af》0.1;
29.e.基因型gt=“het”的样本数》10,且变异比例vaf的最大值《0.2。
30.本发明通过运用多样本联合识别的结果vcf文件中的信息,在不依赖其他数据库和变异注释软件的前提下,运行一次bcftools过滤命令,即可过滤掉频繁出现的假阳性位点以及多态性变异位点,充分利用样本数据自身的质量和频率信息,显著降低了下一步分析过程中需要分析的无效位点个数。本发明通过预先构建变异黑名单,实现不依赖变异注释软件,运行一次bcftools过滤命令,即可对多个样本的变异位点进行过滤,减少了计算机的资源的消耗,缩短了运行时间,在过滤掉良性和高频变异的同时,避免出现假阴性的致病变异位点。
31.本发明在变异黑名单构建环节中,挑选变异分类数据库clinvar中clinsig字段为pathogenic,likely_pathogenic和uncertain_significance的变异,作为白名单,防止在黑名单过滤环节漏掉可能致病的变异。
附图说明
32.图1为本发明构建变异黑名单的流程示意图;
33.图2为本发明变异过滤方法的流程图。
具体实施方式
34.下面结合具体实施例对本发明作进一步描述,但本发明的保护范围并不仅限于此。
35.本发明涉及的相关名词解释如下:
36.acmg指南:特指acmg(the american college of medical genetics and genomics)于2015年3月发表在genetics of medicine杂志的人类基因组变异分类指南,详见参考文献1
37.gnomad:英文全称the genome aggregation database,是一个学术联盟组织,收集和整理了各种大规模的外显子和全基因组测序数据,并面向全世界免费开放。
38.clinvar:一个开放的基因组变异数据库,每个研究机构都可以向其提交数据,对于提交的信息,会有专家团队进行审核评级,对于数据库中的位点,根据注释信息的可靠性,分成了1到4个不同的星级,星级越高,可信度越高。
39.vcf:英文全称the variant call format,是用来存储变异位点信息的标准文件格式。
40.bcftools:一个用于处理变异相关文件(如vcf和vcf文件)的软件。
41.vaf:变异比例,英文全称variant allele fraction,,数值范围是[0,1],计算方式为vaf=depth(alt)/depth(alt+ref),即:变异比例=该位点发生变异的序列数/覆盖该位点的序列总数,当为杂合变异时,vaf越接近0.5代表质量越高,越接近0代表质量越低;当为纯合变异时,vaf越接近1代表质量越高,越接近0代表质量越低。
[0042]
本发明提出了基于多样本全外显子测序的变异过滤方法,其整体流程包括如下步骤:
[0043]
1)构建变异黑名单;
[0044]
2)选取频率阈值和质量阈值,过滤低质量和高频变异;
[0045]
3)根据黑名单进行变异过滤;
[0046]
4)对步骤3)所得结果进行信息注释;
[0047]
5)对步骤4)所得结果按样本进行拆分,得到每个样本注释后的结果。
[0048]
参考附图1,本发明实施例中,构建变异黑名单的具体步骤包括:
[0049]
1.下载gnomad数据库提供的wes的vcf文件,采用bcftools工具提取其中filter字段为pass,且人群频率大于0.02的变异位点,生成“临时黑名单1”;
[0050]
2.下载clinvar数据库提供的vcf文件,采用bcftools工具提取clnrevstat(审核状态)字段为practice_guideline或reviewed_by_expert_panel,且clinsig字段为benign或likely_benign的位点,生成“临时黑名单2”;
[0051]
3.对“临时黑名单1”和“临时黑名单2”中的位点取并集,生成“临时黑名单3”;
[0052]
4.提取clinvar数据库中clinsig字段(致病性结论)为pathogenic,likely_pathogenic和uncertain_significance的位点生成“白名单4”;
[0053]
5.将“临时黑名单3”与“白名单4”取差集,生成vcf格式的“黑名单5”。
[0054]
在本发明的一个可选实施例中,所述步骤2)包括如下步骤:
[0055]
2.1选取恰当的频率阈值:挑选500例回顾性阳性样本,按6例,15例,50例不同样本数梯度从中选择多个样本子集,为各梯度设置不同的变异检出频次阈值,将阳性检出率达
到100%对应各子集的各梯度保留的变异个数从小到大排序,选择排在第1个四分位数附近的阈值作为相应梯度的频率阈值;
[0056]
2.2选取恰当的质量阈值:为500例阳性样本设置多个vaf阈值,将阳性检出率达到100%对应的各子集各梯度保留的变异个数从小到大排序,选择排在第1个四分位数附近的阈值作为相应梯度的vaf阈值;
[0057]
2.3采用bcftools工具view函数的-e参数,根据多样本联合识别变异信息,过滤掉变异质量低于阈值,覆盖深度低于阈值,在本次分析的样本中检出频次高于相应阈值的变异,输出“过滤结果1”。
[0058]
以下结合一个具体实施例对本发明做进一步说明。参考附图2,对100例样本的wes联合识别变异数据进行分析,具体步骤是:
[0059]
一、过滤低质量和高频变异:
[0060]
采用bcftools软件的view函数,判断这100例样本的wes联合识别变异数据“vcf文件1”中的变异位点若符合以下任一条件,则删除,输出“过滤结果1”:
[0061]
a.每个样本的dp(测序覆盖深度)的最大值《10;
[0062]
b.an(等位总数)》12且af(变异频率)》0.5;
[0063]
c.an(等位总数)》30且af(变异频率)》0.2;
[0064]
d.an(等位总数)》100且af(变异频率)》0.1;
[0065]
e.gt(基因型)=“het”的样本数》10,且vaf(变异比例)的最大值《0.2。
[0066]
二、根据变异黑名单进行变异过滤:
[0067]
采用bcftools软件的isec函数,将“过滤结果1”中的变异列表与前述步骤构建的“黑名单5”中的变异列表取差集,输出“过滤结果2”[0068]
三、变异信息注释:
[0069]
采用变异注释软件vep(ensembl variant effect predictor),对“过滤结果2”进行人群频率、致病性、基因、疾病等信息注释,输出“注释结果1”;
[0070]
四、按样本拆分:
[0071]
采用bcftools软件的view函数的-s参数,对“注释结果1”按样本进行拆分,得到每个样本注释后的结果文件“注释结果2”,用于人工筛选和医学遗传学分析。
[0072]
为了评估本发明的效果,本发明对100例样本的wes联合识别变异数据进行分析,采用现有技术方案和本发明方案在同一台linux服务器上,采用10个进程并行,每个进程设置10个线程的运行方式,先后进行变异过滤环节的分析,并比较了两种方案运行的时间和过滤后的变异位点列表的差异,结果如下表1:
[0073]
表1
[0074]
[0075][0076]
由表1可知,本发明可以减少计算机的资源的消耗,缩短运行时间,在过滤掉良性和高频变异的同时,避免出现假阴性的致病变异位点。
[0077]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应该以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1