噻唑类化合物在制备抗SARS-COV-2新型冠状病毒药物中的应用
噻唑类化合物在制备抗sars-cov-2新型冠状病毒药物中的应用
技术领域
1.本发明属于医药技术领域,具体涉及噻唑类化合物在制备抗sars-cov-2新型冠状病毒药物中的应用。
背景技术:
2.新型冠状病毒肺炎(covid-19)是由严重急性呼吸综合征冠状病毒新型冠状病毒(sars-cov-2)引起的严重威胁人类健康的传染性疾病。sars-cov-2是一种含有包膜的rna病毒,由外层的包膜和内层的核衣壳构成。包膜与细胞膜一样同为磷脂双分子层结构,包含病毒基因组编码的结构蛋白,包括刺突糖蛋白(spike glycoprotein,s蛋白)、包膜蛋白(envelope protein,e蛋白)和膜蛋白(membraneprotein,m蛋白)。核衣壳由核衣壳蛋白(nucleocapsidprotein,n蛋白)和病毒rna组装而成。在核酸和蛋白质合成阶段,冠状病毒的2个功能开放阅读框(open reading frame,orf1a/b),翻译出多聚蛋白1a(polyprotein 1a,pp1a)和多聚蛋白1ab(polyprotein 1a,pp1ab),它们被木瓜蛋白酶样蛋白酶(3c-like protease,plpro)和3-胰凝乳样蛋白酶(3c-like protease,3clpro)分割,产生多个非结构蛋白,包括参与病毒转录和复制的rna依赖性rna聚合酶(rna-dependent rna polymerase,rdrp)。
3.sars-cov-2 3clpro和plpro蛋白是sars-cov-2关键蛋白rdrp成为活性形式的关键蛋白。sars-cov-2 3clpro蛋白通过水解病毒pp1a和pp1ab蛋白,使其形成成熟产物nsp1-16,进而组装成具有活性的rdrp。plpro可特异性识别并剪切双甘氨酸多肽,多聚蛋白ppla(pp1ab)n端nsp1-2、nsp2-3和nsp3-4之间的lxgg序列,参与1a(1ab)复制酶蛋白n端的切割加工并释放成熟产物nsp1、nsp2和nsp3,进而组装成具有活性的rdrp。sars-cov-2plpro和3clpro在病毒复制周期起到关键的作用,已被认为是治疗冠状病毒的关键药物靶标。目前,缺乏有效的抗sars-cov-2新型冠状病毒药物。
技术实现要素:
4.本发明提供了通式(i)所示结构的噻唑类化合物,该类化合物是一类抗sars-cov-23clpro蛋白的全新抑制剂,是抗sars-cov-2新型冠状病毒药物先导物的全新分子骨架,可以实现阻断核酸和蛋白质合成的目的,为预防和/或者治疗sars-cov-2新型冠状病毒提供新思路、新用途,具有重要的医学研究价值。
5.本发明一个方面提供如下通式i所示的噻唑类化合物或其药学上可接受的盐在制备抗sars-cov-2新型冠状病毒药物中的应用,
[0006][0007]
其中,
[0008]
r1为一取代至三取代,选自:h,-oh,卤素(f、cl、br、i),c1-c6烷基,c1-c6烷氧基,取代或未取代的氨基-nr3r4,取代或未取代的酰胺基-nh-c(o)-r5,取代或未取代的氨基酰基-c(o)-n-r6r7;
[0009]
r2选自取代或未取代的氨基-nr8r9,取代或未取代的5-7元杂环基酰胺基-nh-c(o)-r
10
,取代或未取代的氨基酰基-c(o)-n-r
11r12
;
[0010]
r3、r4、r8、r9分别独立选自h、取代或未取代的c1-c6烷基,烷基的取代基选自:取代或未取代的芳基、杂芳基,c3-c6环烷基;芳基、杂芳基的取代基为一取代至三取代,选自-oh、卤素(f、cl、br、i),c1-c6烷基,c1-c6烷氧基、c1-c6烷氧基羰基;
[0011]
r5、r
10
分别独立选自取代或未取代的5-7元杂环基、芳基、杂芳基;杂环基、芳基、杂芳基上的取代基为一取代至三取代,选自:-oh、c1-c6烷基、c1-c6烷氧基、c1-c6烷氧基羰基、卤素(f、cl、br、i);
[0012]
r6、r7、r
11
、r
12
分别独立选自取代或未取代的芳基、苯并噻二唑基;芳基、苯并噻二唑基的取代基为一取代至三取代,选自:-oh、c1-c6烷基、c1-c6烷氧基、c1-c6烷氧基羰基、卤素(f、cl、br、i);
[0013]
所述杂环基或杂芳基包含选自n、o、s中的一至三个杂原子;
[0014]
为碳碳键或者碳氢键;
[0015]
n为2时,a为六元环;或者n为1时,a为五元环;或者n为0时,a不成环。
[0016]
在本发明的一种实施方式中,芳基为苯环或者萘环。
[0017]
本发明还提供一种通式i所示的噻唑类化合物或其药学上可接受的盐在制备sars-cov-2 3clpro蛋白抑制剂中的应用。
[0018]
本发明还提供一种预防或治疗sars-cov-2新型冠状病毒肺炎感染的药物,所述药物中含有通式i所示的噻唑类化合物或其药学上可接受的盐、药用辅料。
[0019]
所述药用辅料包括如下任意一种或多种:溶剂、抛射剂、增溶剂、助溶剂、乳化剂、着色剂、黏合剂、崩解剂、润滑剂、润湿剂、渗透压调节剂、稳定剂、助流剂、矫味剂、防腐剂、助悬剂、包衣材料、芳香剂、抗黏合剂、整合剂、渗透促进剂、ph值调节剂、缓冲剂、增塑剂、表面活性剂、发泡剂、消泡剂、增稠剂、包合剂、保湿剂、絮凝剂与反絮凝剂、助滤剂以及释放阻滞剂。
[0020]
在本发明的一种实施方式中,所述药物还包括药物载体。所述药物载体包括微囊、微球、纳米粒和脂质体。
[0021]
在本发明的一种实施方式中,所述药物的剂型包括如下任意一种:注射液、注射用冻干粉针、混悬剂、植入剂、栓塞剂、胶囊剂、片剂、丸剂和口服液。
[0022]
本发明还提供通式i所示的噻唑类化合物的合成方法:
[0023]
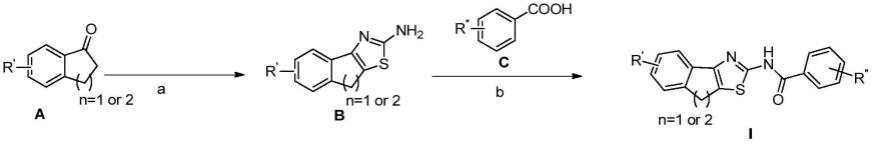
a、当n=1或2,
[0024][0025]
试剂和条件:a)液溴,硫脲,无水乙醇,100℃,5h;b)hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯),dipea(n,n-二异丙基乙胺),dmf(n,n-二甲基甲酰胺),室温,4h;
[0026]
称取a,液溴,硫脲溶解在无水乙醇中,油浴100℃下反应5h,在反应过程中溶液先澄清后变浑浊析出白色固体,tlc监测反应完全后,减压蒸馏除去1/4体积的溶剂,用5%的naoh溶液调碱,抽滤,固体为化合物b;将化合物b,化合物c,hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)溶于dmf(n,n-二甲基甲酰胺)中,滴加dipea(n,n-二异丙基乙胺),室温搅拌4h,监测反应完全后,将反应液滴入水中,析出固体,抽滤干燥,得化合物i;
[0027]
b、当n=0,
[0028][0029]
试剂和条件:a)碘单质,硫脲,110℃,10h;b)hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯),dipea(n,n-二异丙基乙胺),dmf(n,n-二甲基甲酰胺),室温,4h;
[0030]
称取a’,i2,硫脲,油浴110℃熔融状态下反应10h,tlc监测反应完全后,用甲基叔丁基醚打浆后的固体加入水中,用25%的氨水调ph为9-10,抽滤,固体用乙酸乙酯溶解,用饱和nahco3溶液洗涤2-3次,无水硫酸钠干燥有机相,减压蒸馏得化合物b’,将化合物b’,化合物c’,hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)溶于dmf(n,n-二甲基甲酰胺)中,滴加dipea(n,n-二异丙基乙胺),室温搅拌4h,监测反应完全后,将反应液滴入水中,析出固体,抽滤干燥,得化合物i。
[0031]
本发明有益效果:
[0032]
本发明通式(i)所示结构的噻唑类化合物,该类化合物是一类抗sars-cov-2 3clpro蛋白的全新抑制剂,是抗sars-cov-2新型冠状病毒药物先导物的全新分子骨架,可以实现阻断核酸和蛋白质合成的目的,为预防和/或者治疗sars-cov-2新型冠状病毒提供新思路、新用途,具有重要的医学研究价值。
具体实施方式
[0033]
实施例1噻唑类化合物的制备
[0034]
合成方法1、n=1或2
[0035][0036]
试剂和条件:a)液溴,硫脲,无水乙醇,100℃,5h;b)hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯),dipea(n,n-二异丙基乙胺),dmf(n,n-二甲基甲酰胺),室温,4h;
[0037]
称取a,液溴,硫脲溶解在无水乙醇中,油浴100℃下反应5h,在反应过程中溶液先澄清后变浑浊析出白色固体,tlc监测反应完全后,减压蒸馏除去1/4体积的溶剂,用5%的naoh溶液调碱,抽滤,固体为化合物b;将化合物b,化合物c,hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)溶于dmf(n,n-二甲基甲酰胺)中,滴加dipea(n,n-二异丙基乙胺),室温搅拌4h,监测反应完全后,将反应液滴入水中,析出固体,抽滤干燥,得化合物i。
[0038]
具体合成过程:
[0039]
步骤1
[0040][0041]
称取5-甲氧基-1-茚酮a(1.62g,10.0mmol,液溴(1.76g,11.0mmol),硫脲(2.28g,30.0mmol)溶解在20ml无水乙醇中,油浴100℃下反应5h,在反应过程中溶液先澄清后变浑浊析出白色固体,tlc监测反应完全后,减压蒸馏除去1/4体积的溶剂,用5%的naoh溶液调碱,抽滤,固体为化合物wj-218(1.8g,产率50%)。
[0042]
步骤2
[0043][0044]
称取化合物wj-218(0.24g,1.1mmol),3-甲氧基苯甲酸(0.15g,1.0mmol),hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)(0.38g,1.0mmol)溶于5ml dmf(n,n-二甲基甲酰胺)中,滴加dipea(n,n-二异丙基乙胺)(0.39g,3.0mmol),室温搅拌4h,监测反应完全后,将反应液滴入水中,析出固体,抽滤干燥,得化合物wj-354(0.18g,产率50%)。
[0045]
替换相应底物,按照相同方法制备如下化合物。
[0046]
[0047]
[0048]
[0049]
[0050][0051]
合成方案2、n=0
[0052][0053]
试剂和条件:a)碘单质,硫脲,110℃,10h;b)hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯),dipea(n,n-二异丙基乙胺),dmf(n,n-二甲基甲酰胺),室温,4h;
[0054]
称取a’,i2,硫脲,油浴110℃熔融状态下反应10h,tlc监测反应完全后,用甲基叔丁基醚打浆后的固体加入水中,用25%的氨水调ph为9-10,抽滤,固体用乙酸乙酯溶解,用饱和nahco3溶液洗涤2-3次,无水硫酸钠干燥有机相,减压蒸馏得化合物b’,将化合物b’,化合物c’,hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)溶于dmf(n,n-二甲基甲酰胺)中,滴加dipea(n,n-二异丙基乙胺),室温搅拌4h,监测反应完全后,将反应液滴入水中,析出固体,抽滤干燥,得化合物i。
[0055]
具体合成过程:
[0056]
步骤1
[0057][0058]
称取a’(1.64g,10mmol),i2(2.54g,10mmol),硫脲(0.76g,10mmol),油浴110℃熔融状态下反应10h,tlc监测反应完全后,用甲基叔丁基醚打浆后的固体加入水中,用25%的氨水调ph为9-10,抽滤,固体用etoac(50ml)溶解,用饱和nahco3溶液洗涤(20ml
×
3)次,无水硫酸钠干燥有机相,减压蒸馏得化合物wj220(1.10g,产率50%)。
[0059]
步骤2
[0060][0061]
称取化合物wj-220(0.24g,1.1mmol),3-甲氧基苯甲酸(0.15g,1.0mmol),hatu(2-(7-偶氮苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯)(0.38g,1.0mmol)溶于5ml dmf(n,
avlqsgfrk(dnp)k),sars-cov-2 3clpro酶溶液(在反应液中稀释到0.5μm)与化合物在反应液(20mm tris,ph7.3,150mm nacl,1mm edta,1%glycerol,0.01%tween-20)中室温共孵育10分钟,加入底物(40μm,反应液总体积50μl),开始反应后用envision multilabel reader(perkinelmer)检测反应液的荧光强度(激发光320nm,发射光405nm),每个剂量设三个复孔。对照孔(dmso)的荧光值设为100%,化合物处理孔用相对于对照孔的百分数来表示其活性率。
[0067]
8h-茚并[1,2-d]噻唑类化合物以不同浓度(浓度从0.08μm到20μm)对3clpro蛋白酶活抑制作用进行检测,测试活性剂量依赖关系,即ic
50
/ec
50
值,通过样品活性对样品浓度进行非线性拟和得到,计算所用软件为graphpad prism 8,拟合所使用的模型为四参数剂量效应积分模型(four-parameter concentration
–
response model)(varible slope),对于大多数抑制剂筛选模型,将拟合曲线底部和顶部设定为0和100。一般情况下,每个样品在测试中均设置复孔(n≥3),在结果中以标准偏差(standard deviation,sd)或者标准误差(standard error,se)表示。计算ic
50
。
[0068]
实施例3 8h-茚并[1,2-d]噻唑类化合物对sars-cov-2plpro蛋白酶活抑制作用检测:
[0069]
基于sars-cov-2plpro蛋白是一种蛋白水解酶的基本特点,建立荧光法检测sars-cov-2plpro蛋白活性的筛选体系。sars-cov-2plpro蛋白可特异性识别并剪切双甘氨酸多肽,其活性检测可采用荧光多肽为底物,通过检测荧光信号的生成来反应其蛋白水解酶的活性。sars-cov-2plpro蛋白酶活检测使用香豆素标记的多肽底物(序列:z-rlrgg-amc),sars-cov-2plpro酶溶液(在反应液中稀释到40nm)与化合物在反应液(20mm tris ph8.0,0.01%tween20,0.5mm dtt)中室温共孵育10分钟,加入底物(50μm,反应液总体积50μl),开始反应后用envision multilabel reader(perkinelmer)检测反应液的荧光强度(激发光355nm,发射光460nm),每个剂量设三个复孔。对照孔(dmso)的荧光值设为100%,化合物处理孔用相对于对照孔的百分数来表示其活性率。
[0070]
8h-茚并[1,2-d]噻唑类化合物以不同浓度(浓度从0.08μm到20μm)对plpro蛋白酶活抑制作用进行检测,测试活性剂量依赖关系,即ic
50
/ec
50
值,通过样品活性对样品浓度进行非线性拟和得到,计算所用软件为graphpad prism 8,拟合所使用的模型为四参数剂量效应积分模型(four-parameter concentration
–
response model)(varible slope),对于大多数抑制剂筛选模型,将拟合曲线底部和顶部设定为0和100。一般情况下,每个样品在测试中均设置复孔(n≥3),在结果中以标准偏差(standard deviation,sd)或者标准误差(standard error,se)表示。计算ic
50
。
[0071]
五、试验方法
[0072]
(1)细胞培养和处理:
[0073]
vero e6细胞培养于t75的细胞培养瓶中,置于37℃,5%co2的培养箱培养,每48小时按1:3比例进行传代,培养基配方:90%dmem(gibco invitrogen),10%胎牛血清(gibco invitrogen)。
[0074]
测试前一天,吸走细胞培养液,用细胞外液淋洗一遍后加入2ml trypsin溶液,在室温下消化1-2分钟。加入培养基中和胰酶,对细胞进行计数,并将细胞传到48孔板中,每孔50000个细胞。
[0075]
(2)化合物准备:
[0076]
测试化合物分别用dmso溶解成40mm母液,测试当天,将化合物母液用dmem进行10倍连续稀释,即取1μl的化合物母液加入到9μl dmso中,经过2个10倍稀释后,得到0.4mm的稀释液后,40倍稀释至10μm。
[0077]
阳性对照化合物磷酸氯喹的准备,化合物用pbs溶解成2mm母液。
[0078]
化合物最终测试浓度中dmso的含量不超过0.2%,此浓度的dmso对sars-cov-2复制没有影响。
[0079]
(3)检测化合物对病毒复制的影响
[0080]
48孔板的细胞移除上清,每孔加入稀释好的化合物,孵育1小时,在生物安全3级(bsl-3)实验室里加入sars-cov-2,感染复数(moi)为0.01,孵育1小时后,移除上清,pbs洗,加入稀释好的化合物,感染后24小时去掉上清,孔板细胞用多聚甲醛固定。细胞固定后,先后用sars-cov-2np蛋白兔多抗、488荧光抗体和dapi染料进行孵育、染色,将48孔板置于荧光显微镜成像系统下进行自动扫描拍照,根据dmso孔的病毒载量(绿色荧光)和细胞数量(蓝色荧光)为对照,利用image j软件对荧光信号进行统计计数,分别计算化合物抑制率和药物毒性。
[0081]
(4)数据处理及统计
[0082]
数据分析处理采用excel软件,以下公式计算:
[0083]
inhibition%=[1
–
(i/io)]
×
100%
[0084]
其中,inhibition%代表化合物对sars-cov-2复制的抑制百分率,i和io分别表示在化合物和对照孔(dmso组)sars-cov-2病毒rna在细胞中的荧光信号面积。
[0085]
cytotoxicity的计算方法同上。
[0086]
(5)实验结果
[0087]
上述实验过程获得的测试结果见表1。
[0088]
表1
[0089]
[0090]
[0091]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1