一种基于转录组快速获取无基因组物种目标基因家族的方法
本发明涉及转录组测序数据分析领域,具体而言,涉及一种无基因组物种的转录组分析方法,尤其涉及一种基于二代测序技术获得的转录组数据,快速获取无基因组物种的目标基因家族的方法。
背景技术:
1、长期以来,一代测序技术一直是分子生物学相关研究中最常用的技术手段之一,通过设计引物,使用pcr克隆来进行测序,从一定程度上推动了该领域的快速发展,但是其存在测试速度慢、成本高、通量低等方面的不足。二代测序技术是基于边合成边测序的原理开发的测序技术,又称大量并行测序技术、高通量测序技术,以低成本、99%以上的准确度,1次可对几百、几千个样本的几十万至几百万条核苷酸分子同时进行快速测序分析,是对第一代测序技术的划时代变革的核心,缺点是测序度长短,普遍在20-300bp,样本制备过程繁琐。三代测序技术是基于单分子测序技术原理开发的测序技术,优点是超长读长,缺点是测序准确率较低。
2、转录组测序是指细胞或组织的特定状态下,能转录出来的所有rna的总和,主要包括mrna和非编码rna,可以研究样品间转录水平的差异表达。转录组数据可以通过二代或三代测序获得。三代测序虽然可以更高效地获得片段更长的转录组数据,但由于其价格昂贵,经济压力大,而且准确率不高,影响了普及率,因此主要还是通过二代测序来获取转录组数据。
3、对于无基因组物种的目标基因家族序列的获取,通常有两种方法:一、查找模式物种基因序列,采用一代测序方法,设计简并引物,使用pcr克隆,测序方式获取;二、通过二代测序获得转录组等高通量测序数据,参照测序公司给予的注释信息直接检索。
4、第一种方法的实验需要试剂、设备和较长时间,较多不确定性,成本较高,而结果获取较为困难;第二种方法由于测序公司给予较多的注释信息,同时常用的7大数据库,如nr数据库,由于其开源的方式虽然数据量较多,但存在较多的错误注释信息,kegg虽然较为准确但仅仅研究较多的基因或蛋白有注释,其他数据库存在问题更多,且生信分析人员通常缺乏对某一特定研究对象的专业知识,通过常规参数获得结果指向性较差,如何高效、准确、全面的获取目标基因家族是系统性研究家族基因的难点。
5、对于无基因组(目前除模式植物外,大多数物种均不存在基因组)的植物而言,系统性研究其某一功能基因家族的前提是获取到全面的基因家族信息,而如何系统性的获取家族中所有类别的基因或蛋白序列是一线研究人员的研究起始。
6、cn112397149提供了一种无参考基因组序列的转录组数据进行分析的方法及系统,主要通过r语言等软件平台及注释信息进行分析。
7、但是生物专业的研究人员往往由于缺乏对转录组或基因组数据处理等方面的计算机领域专业知识 (如r语言等软件平台、运算代码撰写)或缺乏运算数据所需高质量设备,无法直接从转录组(特别是无参的转录组)中快捷的获取准确信息。
8、同时生信分析人员中大多数人员其专业背景为计算机相关专业背景,往往不具备具体到某一功能的基因或蛋白层面的生物学专业知识储备,其处理数据往往利用公开数据库的信息进行算法或参数优化筛选目标基因,针对某一基因家族等精确性研究其结果往往存在较多的错误和遗漏,同时无参转录组其在组装和注释过程中,基因家族成员由于序列的相似度较高,极易造成结果中序列不完整、组装错配、注释错误等情况。
9、因此急需找到一种既能适用于生物专业的研究人员、又能适用于生信分析人员的,不需要专业的生物信息学知识,也无需使用r语言等软件平台、运算代码撰写,能够更加简便、准确、快速的从转录组数据获得无基因组物种的目标基因家族的所有序列的方法。
技术实现思路
1、为解决上述问题,本发明提供了一种基于转录组快速获取无基因组物种的目标基因家族的方法,通过选取参照物种、系列模式物种,并基于转录组数据,组合使用基因搜索比对软件bioedit、进化树构建软件mega6和在线分析软件meme,构建了一种可视化、不需要专业的生物信息学知识,也无需使用r语言等软件平台、运算代码撰写的,针对无基因组物种,快速、高效、准确、全面地获取目标基因家族的所有序列的方法,成功获得杭白菊的pebp基因家族所有序列,并经一代测序验证,准确率为100%。
2、目前获取无基因组物种的目标基因序列,主要通过7大数据库,如nr数据库,由于其开源的方式虽然数据量较多,但存在较多的错误注释信息,kegg虽然较为准确但仅仅研究较多的基因或蛋白有注释,其他数据库存在问题更多。
3、本发明提供的基于转录组快速获取无基因组物种的目标基因家族的方法,不直接使用目标转录组的注释信息,从而避开生物信息技术或公开数据库错误等原因造成的错误信息,流程中使用的所有软件均为可视化的生物信息学软件或在线网站,所需生物信息学专业知识较少,基于少量准确的文献信息即可获得完成序列的搜索和鉴定,可使一线研究人员较快上手。
4、一方面,本发明提供了一种快速获取无基因组物种的目标基因家族的方法,所述方法包括以下步骤:
5、(1)选取参照物种、系列模式物种,利用参照物种、系列模式物种的全基因组数据,以及待测物种的转录组数据,通过bioedit软件构建系列模式物种和待测物种的目标基因家族本地数据库;所述待测物种为无基因组物种;
6、(2)通过mega6软件构建进化树,使待测物种的目标基因家族本地数据库中的氨基酸序列利用进化树的排序规则进行分类;
7、(3)利用meme软件进行motif分析;
8、(4)氨基酸序列单独比对。
9、本发明所述系列模式物种,是指针对目标基因家族选取的,包括不同进化阶段、不同形状特征等的相关物种。
10、本发明所述bioedit软件为美国borland公司基于borland c++buider 3.0编写开发的一款分子生物学分析软件,支持序列比对、序列检索、引物设计等功能,能够用于完成核苷酸序列或氨基酸序列的所有常规分析操作。该软件为可视化生信软件,仅需部分简单的生物学知识就可操作,不需要涉及语言、编程等计算机专业知识。
11、在一些方式中,本发明采用的bioedit软件的具体版本为bioedit 7.2.6.1。
12、本发明所述mega6(molecular evolutionary genetics analysis)软件是一个分子进化遗传分析软件,可用于序列比对、进化树的推断、估计分子进化速度、验证进化假说,可以编辑序列数据、序列比对、构建系统发育树、推测物种间的进化距离等。该软件为可视化生信软件,仅需部分简单的生物学知识就可操作,不需要涉及语言、编程等计算机专业知识。
13、在一些方式中,本发明采用的mega6软件的具体版本为mega 6.06。
14、本发明所述meme(motif-based sequence analysis tools)是meme suite软件套件中用于检索motif (功能特征序列)的工具,motif是比较有特征的短序列,可能会多次出现的,并被假设拥有生物学功能。该软件为可视化生信软件,仅需部分简单的生物学知识就可操作,不需要涉及语言、编程等计算机专业知识。
15、在一些方式中,本发明采用的meme软件的具体版本为meme 5.4.1。
16、在一些方式中,所述目标基因家族为pebp(磷脂酰乙醇胺结合蛋白,phosphatidylethanolamine-binding proteins)基因家族。
17、在一些方式中,需要针对pebp基因家族来选取系列模式物种,考虑到pebp基因功能以花芽分化和花器官发育为主,因此系列模式物种考虑选择无花植物、双子叶植物、单子叶植物中多年生和一年生植物,同时还可以根据pebp基因家族的进化程度来选取可能处于不同进化进程的植物。
18、在一些方式中,所述待测物种为杭白菊。杭白菊目前在公开网站都尚未有基因组报道,本发明通过二代测序获取杭白菊的转录组数据,并针对pebp基因家族选取合适的参照物种和系列模式物种,再经组合使用基因搜索比对软件bioedit、进化树构建软件mega6和在线分析软件meme,成功获得杭白菊的pebp基因家族所有序列,并经一代测序验证,准确率为100%。可见采用本发明提供的方法可以非常低的成本(二代测序单一样本数据库测序加组装建库仅需600-800元),快速精准地获取无基因组物种的目标基因家族所有序列,相比一代测序节约了大量的人力、物力和财力。
19、在一些方式中,所述待测物种为茶树。
20、在一些方式中,所述目标基因家族为gaox氧化酶基因家族(包括ga20氧化酶、ga3氧化酶和 ga2氧化酶)。
21、可以理解的是,针对任何目标基因家族,任何无基因组的物种,采用本发明提供的方法,都可以低成本且快速精准地获取无基因组物种的目标基因家族所有序列,而且全程可视化,不需要专业的生物信息学知识,也无需使用r语言等软件平台、运算代码撰写,因此都在本发明的保护范围内。
22、进一步地,步骤(1)还包括确定参照物种的目标基因家族的成员类型;所述系列模式物种包括至少十种具有全基因组数据,且都具有目标基因家族的物种;所述参照物种为系列模式物种中的一种。
23、在采用本发明提供的方法,在开始步骤(1)之前,最好能先做好充足的准备工作,包括针对目标基因家族,先查阅大量公开文献资料,了解该目标基因家族的一些信息。
24、在一些方式中,针对pebp基因家族,可以先查阅相关公开发表文献,了解哪些物种在pebp基因家族方面研究较多,有较完善的基因组,从而确定可以选取拟南芥、水稻等基因组较完善的研究对象作为参照物种,再综合多篇文献确认拟南芥或水稻的pebp基因家族成员类型(包括ft、tfl1、mft等),结构域为pf00161,同时下载文献作者上传的对应序列供参考。
25、在一些方式中,经查阅公开发表文献,pebp基因家族可分为ft-like、tfl-like和mft-like三大类别。
26、在一些方式中,本发明选取拟南芥作为参照物种。
27、在一些方式中,本发明经查询拟南芥tair数据库,在线网站,以关键词pebp、ft、tfl1、mft、 pf00161等pebp基因家族相关信息,检索pebp基因家族成员,确定拟南芥的pebp基因家族共有6 个成员(也就是6条蛋白序列):atft(at1g65480)、attsf(at4g20370)、attfl1(at5g03840)、 atatc(at2g27550)、atbft(at5g62040)和atmft(at1g18100)。
28、在一些方式中,针对pebp基因家族功能以花芽分化和花器官发育为主,模式物种可以选取双子叶植物、单子叶植物中多年生和一年生植物,同时根据进化进程选择pebp基因家族不同进化阶段的植物,还可以选择进化程度较低的无花植物(如江南卷柏),藓类植物(如小立碗藓);因此为了获取杭白菊的pebp基因家族序列,选取系列模式物种如下:拟南芥arabidopsis、克莱门柚citrus clementina、橙子citrus sinensis、黄瓜cucumissativus、大豆glycine max、紫荆苜蓿medicago truncatula、水稻oryza sativa、小立碗藓physcomitrella patens、杨树populus trichocarpa、桃prunus persica、江南卷柏selaginella moellendorffii、高粱sorghum bicolor和玉米zea mays,共计13个物种,其中作为参照物种的拟南芥也是系列模式物种中的一种。
29、在一些方式中,为了获取茶树的gaox氧化酶基因家族,考虑到gaox氧化酶基因家族的性质,选取参照物种为拟南芥,选取系列模式物种为拟南芥arabidopsis、橙子citrussinensis、黄瓜cucumis sativus、大豆glycine max、紫荆苜蓿medicago truncatula、小立碗藓physcomitrella patens、杨树populus trichocarpa、江南卷柏selaginellamoellendorffii、高粱sorghum bicolor、葡萄vitis vinifera和玉米zea mays,共计11个物种。
30、进一步地,步骤(1)所述通过bioedit软件构建本地数据库,包括通过bioedit软件的blastp程序从系列模式物种的基因组数据中获取目标基因家族的氨基酸序列及相应核苷酸序列,通过bioedit软件的 blastx和blastp程序从待测物种的转录组数据中获得目标基因家族的氨基酸序列及相应核苷酸序列。
31、本发明通过bioedit软件来全面搜索参照物种、系列模式物种的完整基因组数据,从中获取更全的目标基因家族所有序列。
32、在一些方式中,由于氨基酸序列直接决定蛋白结构和功能,本发明主要通过构建完整氨基酸序列数据库,通过氨基酸序列来确定基因家族数据。
33、对于无基因组数据的待测物种,则主要通过转录组数据来翻译和构建完整氨基酸序列数据库。
34、所述blastp程序可用于搜索和比对蛋白序列;所述blastx程序可用于实现核酸序列到氨基酸序列的翻译和比对;因此可通过blastp来搜索和比对参照物种、系列模式物种的氨基酸序列,可通过blastx程序来获取待测物种的氨基酸序列,并可通过blastp来辅助搜索待测物种的氨基酸序列。
35、进一步地,所述使用blastp程序时,需设置参数expectation value为10-3,设置同源性为40%,搜索结果还需去重,使每条氨基酸序列只保留唯一编号。
36、依次将所有序列设为“查询目标(query)”进行比对,查看获得的结果,选取标准identities超40%以上,将比对的获得的目标序列作为备选序列,记录对应的基因组编号。
37、使用blastp程序进行氨基酸序列搜索时,设置较高的expectation value和较低的同源性,能搜索到更多的氨基酸序列。经过多次试验证明,通过设置参数expectationvalue为10-3,设置同源性为40%,能够最大程度地获取更多的氨基酸序列,从而保证获得全面完整的目标基因家族的所有氨基酸系列。当设置参数expectation value为10-4,或是设置同源性为80%,获得的氨基酸序列会明显减少,从而有可能导致难以获得全面的基因家族数据。
38、当设置参数expectation value为10-3,设置同源性为40%时,虽然能够最大程度地获取更多的氨基酸序列,但是由于每采用一条氨基酸序列进行搜索时,会搜索到大量的类似序列氨基酸,会出现获得大量重复氨基酸序列的问题。比如采用拟南芥的atft(at1g65480)成员的一条氨基酸序列设为“查询目标(query)”进行搜索时,可能会搜索到atft(at1g65480)成员的大量氨基酸序列,同时还会搜索到attfl1(at5g03840)等其他成员的大量氨基酸序列;当采用attfl1(at5g03840)成员的一条氨基酸序列设为“查询目标(query)”进行搜索时,也可能会搜索到atft(at1g65480)成员的大量氨基酸序列,同时还会搜索到atft(at1g65480)等其他成员的大量氨基酸序列,因此导致整个搜索过程会产生大量的重复氨基酸序列,还需进行序列比对和去重,使每条氨基酸序列只保留唯一基因组编号,使用唯一编号调取数据库中的氨基酸序列。
39、进一步地,通过bioedit软件构建本地数据库后,还需使用pfam31.0进行蛋白结构域的完整度鉴定,使系列模式物种和待测物种的目标基因家族的蛋白结构域达到100%完整。
40、同一个基因家族的每一个成员都会有一个相同的结构域,不同物种的同一个基因家族,同样拥有相同的结构域。这个结构域可以由pfam31.0中的隐马尔可夫模型计算匹配后得到,隐马尔可夫模型是一种计算方式,可用于计算一个基因家族的结构域。根据参考物种的结构域,可用于待测物种的基因家族每个成员的结构域完整性鉴定。
41、通过验证pebp家族的结构域的完整性,可以帮助验证pebp家族的氨基酸系列完整性。
42、在一些方式中,还可以通过smart在线网站来鉴定目标基因家族的结构域的完整性。
43、在一些方式中,还可以通过ncbi在线网站来看相似序列的注释,帮助进一步判断和鉴定氨基酸序列的完整性。
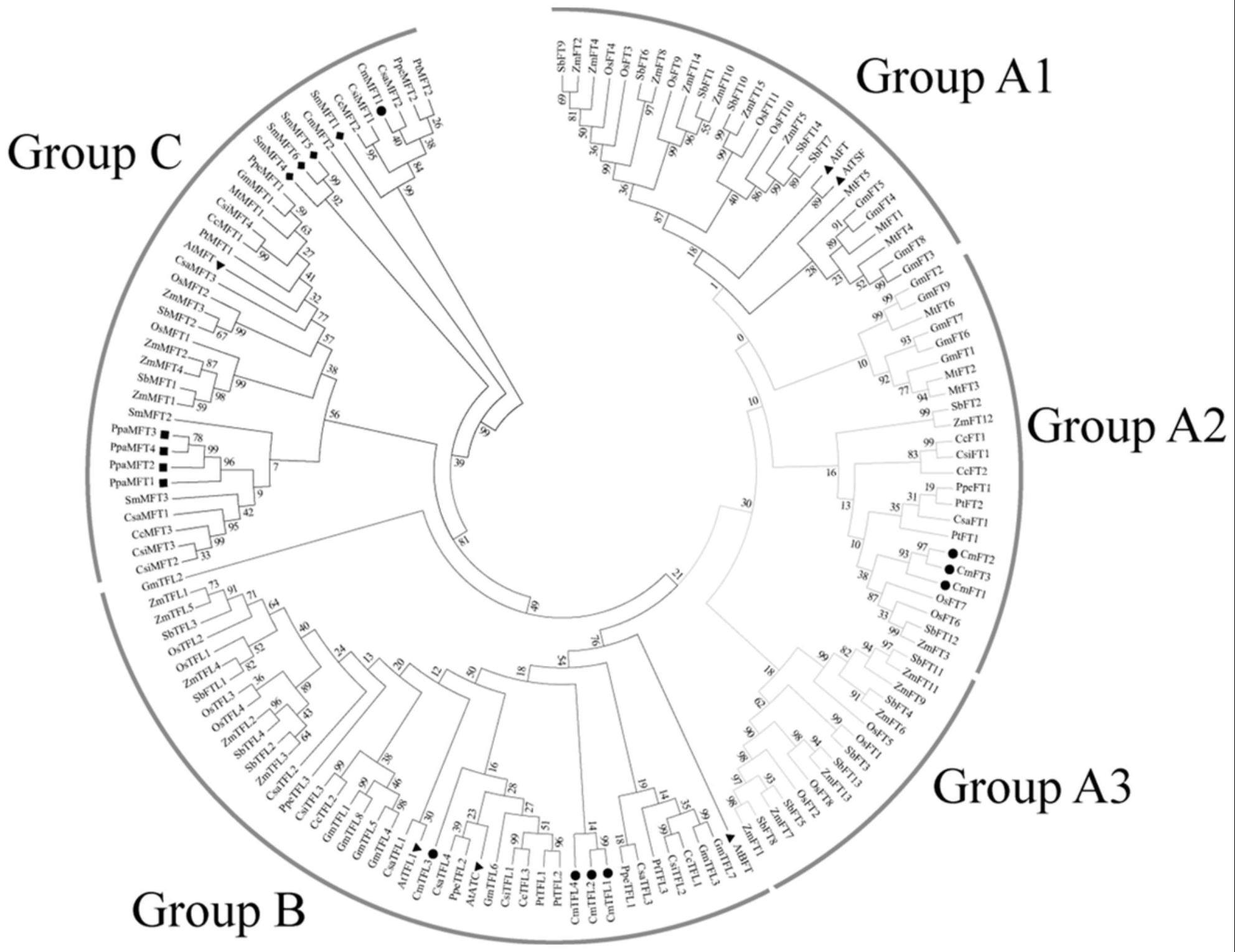
44、值得注意的是,在这一步的鉴定过程中,注意是检查序列的结构域完整性,即使发现有些错误序列、疑似错配,也可以先保留待后面步骤进一步鉴定,不用删除序列。
45、本技术通过bioedit软件找到所有的可能序列,再进入下一步的分类鉴定,能最大程度减少误差。
46、进一步地,步骤(2)所述mega6软件构建进化树,需先导入系列模式物种和待测物种的氨基酸序列,选择phylogeny中neighbor-joining tree,参数设置bootstrap为1000。
47、bootstrap为自展值,用于检验计算的进化树分支可信度,bootstrap的值越大表示结果越准确,参数设置bootstrap为1000,已经足够满足本发明构建进化树的需求,可以获得可信度非常高的进化树。
48、通过mega6软件构建进化树,可以对其中的所有氨基酸序列进行聚类分析,越相似的氨基酸序列会被聚类到更近一些的分叉位置,越不相似的序列则会被分到更远一些的分叉位置。
49、mega6软件构建进化树,通常是用于查看新获得基因序列与其同类型基因序列来构建进化树,查看基因处于进化树的哪一个进化阶段。而本技术巧妙地将mega6软件构建进化树的功能,用于基因家族内部的分类和去重复,根据参照物种的目标基因家族成员类型,从而帮助确定待测物种的目标基因家族序列分别具有哪些家族成员,也相当于能基本确定该待测物种的目标基因家族具有几条氨基酸序列。
50、进一步地,步骤(2)所述mega6软件构建进化树时,如2条氨基酸序列的分支数值为100且属于同一物种,需进行单独氨基酸序列比对;单独氨基酸序列比对过程中,如2条氨基酸序列相似度超99%,则去掉其中1条,如未达到99%则全部保留。
51、在一些方式中,通过mega6软件构建进化树的基本操作为:先将已检索的13个物种和待测物种的pebp蛋白序列保存于txt文件中;然后在mega6软件中导入序列后,选择alignment中align by clustalw方法的分析结果;点击file,打开保存的xx.mas文件,点击analyze,选择phylogeny中 neighbor-joining tree,参数设置bootstrap为1000;导出结果进行查看。如2个序列的分支数值(可信度)为100且属于同一物种,进行单独序列比对,如2条序列相似度超99%,则去掉其中1条,如未达到99%则保留(认为是2条存在差异的序列)。
52、构建进化树过程中,每一个分支都会提供一个可信度的值,如果两条序列的分支可信度为100,且属于同一物种,则表示这两条序列有可能为完全相同的蛋白序列,需单独进行序列比对,进行单独序列比对时,如果2条序列相似度超99%,则说明这两条为完全相同的重复序列,需要去掉其中1条,如未达到99%则保留,此过程相当于去重的过程。
53、进一步地,删除相似度超99%的氨基酸序列后,剩余氨基酸序列再次导入mega6软件构建进化树。
54、经去重后的氨基酸序列,与所有系列模式物种的氨基酸序列,再次构建进化树,从而对剩余序列进行初步分类。
55、在一些方式中,根据步骤(1)中检索的文献显示,pebp基因家族可分为ft-like、tfl-like和mft-like 三大类别,本次构建进化树,根据拟南芥序列分布进行类别判定,对待测物种杭白菊以及其他系列模式物种的氨基酸序列都进行初步分类。
56、在一些方式中,根据拟南芥的区分,再结合系列模式物种的氨基酸序列构建进化树后,我们可以将整个进化树分为3个大组,5个小组,其中groupa是ft-like,groupb是tfl-like,groupc是mft-like。
57、进一步地,步骤(3)所述的利用meme软件进行motif分析,以参照物种作为参照对象,通过保守 motif来进行鉴定和分类,区分待测物种的目标基因家族内部成员类型。
58、由于经步骤(1)中pfam31.0进行结构域pbp的完整度鉴定,和步骤(2)中进化树的简单分类,还有可能存在将近似的基因当做目标基因,还不能解决转录组序列不完整、组装错误来源数据有误等情况。因此还需要利用meme在线数据库进行motif分析,通过meme,可以对初步分类后的每一类序列进行motif分析,进一步来鉴定和分类,区分基因家族的内部成员,同时排除功能差异较大的序列。
59、基因的功能与其蛋白质一维氨基酸排列顺序密切相关,因此存在较多的保守motif(motif是比较有特征的短序列,可能会多次出现的,并被假设拥有生物学功能),可以此来鉴定和分类,根据其差异可区分基因家族的内部成员。
60、在一些方式中,功能差异较大的序列有可能不是目标序列,有可能是组装错误的基因等等,可以通过motif分析来进行去除。
61、还有一些方式中,功能差异较大的序列也可能是同一个基因家族中的不同成员,比如pebp基因家族中mft基因与其他两类家族成员的差别较大,因此必须专门针对进化树分类获得的每一类进行分析,以防止meme直接排除与其他两类差别较大的mft基因。
62、本发明巧妙地通过先使用mega6软件再使用meme软件,帮助快速分类和鉴定,删除重复和错误序列,从而获得待测物种的目标基因家族序列,整个过程非常便捷高效。
63、进一步地,步骤(4)所述的氨基酸序列单独比对,是指针对在进化树上分类距离接近的氨基酸序列,在motif分析时,应该具有一致的保守motif,否则需单独对比氨基酸序列,检查序列的完整性、排除拼接错误。
64、为了进一步提升pebp成员鉴定结果的准确性和分类单元的准确度,针对根据进化树大类别分析结果,和meme的motif分析的排除错误过程,从而获得的pebp成员,还需进一步从氨基酸排列层面进行比对分析。
65、根据公开发表文献和拟南芥pebp成员在pfam31.0中的domain结构域和保守结合位点进行标注。根据结构域中的保守序列、保守结合位点进行判定,如果来自多个物种的几个基因在进化树上分类距离较近,那么这些基因在序列上的保守序列、保守结合位点呈现一致性或类似性(多条序列呈现一致也是保守的),如出现其他情况,则基因序列可能存在错误、不完整、转录组的组装错误等情况,可选取目标物种中进化树比较接近的基因对象单独比对,检查序列的完整性、排除拼接错误。
66、再一方面,本发明提供了一种软件的组合用于快速获取无基因组物种的目标基因家族的用途,所述软件组合包括bioedit软件、mega6软件和meme在线软件。
67、本发明的有益效果:
68、(1)构建了一种可视化、不需要专业的生物信息学知识,也无需使用r语言等软件平台、运算代码撰写的,针对无基因组物种,快速、高效、准确、全面地获取目标基因家族的所有序列的方法;
69、(2)成功获得杭白菊的pebp基因家族所有序列,并经一代测序验证,准确率为100%。
70、(3)通过结合bioedit软件的序列搜索和比对功能,以及mega6软件和meme软件进行分类、去重,以及最后针对某些单独氨基酸序列的完整性、排除拼接错误比对,以最简单高效的手段快速获得无基因组物种的目标家族基因;
71、(4)解决了传统生物学技术手段和现代生物信息学手段获取基因信息存在的巨大信息差,可以节约传统生物学技术手段的大量时间,校正现代生物信息学手段的限制因素,在非实验的手段下将无参考基因组的杭白菊中的目标基因家族进行全面、快捷的获得;
72、(5)不使用目标转录组的注释信息,从而避开生物信息技术或公开数据库错误等原因造成的信息错误信息,流程中使用的所有软件均为可视化的生物信息学软件或在线网站,所需生物信息学专业知识较少,基于少量准确的文献信息即可获得完成序列的搜索和鉴定,可使一线研究人员较快上手。
- 还没有人留言评论。精彩留言会获得点赞!